Généralités sur les structures de protéines
|
|
|
- Cyril Mongeau
- il y a 10 ans
- Total affichages :
Transcription
1 Généralités sur les structures de protéines Le Paradoxe de Levinthal: En 1969 Cyrus Levinthal a noté qu'à cause du grand nombre de degrés de liberté dans un polypeptide déplié, une protéine a un nombre astronomique de conformations (une estimation de est donnée dans l'article original). Si la protéine cherchait sa conformation repliée correcte séquentiellement, il lui faudrait un temps plus grand que l'âge de l'univers. Ceci est vrai même si les conformations étaient testées à un taux rapide (ordre de la picoseconde). En effet, si l'on considère seulement 2 états de conformation pour un acide aminé, un polypeptide de 100 acides aminés possèderait états possibles de conformation (soit environ ). Si on donne 1 picoseconde (10-12 s) pour changer de conformation, il faut *10-12 soit secondes ou encore ans. Beaucoup de petites protéines se replient spontanément sur une échelle de temps de l'ordre de la milliseconde ou même de la microseconde. Le temps de génération de E. coli peut être de l'ordre de 20 mn (phase exponentielle de croissance), ce qui indique que toutes les protéines essentielles se replient sur un temps d'au plus quelques minutes. En fait, le paradoxe montre simplement que les protéines se replient en suivant un chemin non aléatoire qui n'explore pas toutes les possibilités. De plus, les conformations d'un acide aminé dépendent de celles de ses voisins (l'indépendance suposée dans le paradoxe est fausse). Quelques faits : Le rapport entre résidus hydrophiles et hydrophobes est à peu près constant (h/h=0.36 écart type:0.06). Les protéines enfouissent une fraction constante de leur surface totale - C & S sont enfouis à 86% - O & N neutres sont enfouis à 40% - O & N chargés sont enfouis à 32% L intérieur des protéines est aussi dense que des cristaux de petites molécules organiques Type Densité d'empaquetage % Liquides Solides organiques Glace 58 Protéines La stabilité de la forme repliée est marginale (5-10 kcal/mol) Les forces qui stabilisent la forme repliée: Liaisons hydrogènes intramoléculaires Entropie de déshydratation Interactions hydrophobes Ponts salins Interactions dipôlaires Les forces qui déstabilisent la forme repliéé Liaisons hydrogènes avec l eau Perte d entropie configurationnelle Méthodes de modélisation moléculaire voir cours
.")
2 Contexte: En 2006, il y a dans la banque cristallographique: fichiers PDB (13000 en 2000, 3800 en 1995) Si on regroupe les protéines par groupes basés sur l'identité de séquence, on constate la redondance de la PDB. % identité Nombre de Clusters 95% identité % identité % identité % identité Sur les nouvelles structures de protéines trouvées: - 1/3 sont similaires (ou homologues) à des protéines connues - 1/4 sont similaires à une protéine de structure connue Il existe des structures 3D similaires dont les séquences primaires divergent (<15% d'identité). Note: 15% d'indentité est la "ligne de base" pour un alignement, c'est à dire que 2 protéines complètement différentes ne peuvent avoir moins de 15% d'identité après alignement. Classification SCOP La classification SCOP (Murzin et al., 1995; Conte et al., 2002; Brenner et al., 1996) est faite manuellement d après des informations structurales et des connaissances plus générales sur chaque protéine. Les outils automatiques de comparaison structurale ne sont utilisés que pour aider à la classification par inspection visuelle. Les structures protéiques sont tout d abord découpées en domaines (régions ayant un coeur hydrophobe et peu d interaction avec le reste de la protéine) puis sont classées. Les quatre niveaux de classification sont, du niveau le plus général au plus fin : 1. class : la composition en structures secondaires est similaire. Il y a quatre classes principales qui sont les mêmes que celles déjà citées et définies par M. Levitt et C. Chothia (Levitt and Chothia,1976). Les 7 autres classes ont un effectif beaucoup plus faible. Les classes des protéines multidomaines, des protéines membranaires et des petites protéines sont de vraies classes où les protéines ont des caractéristiques spécifiques tandis que les autres sont plus des artefacts dus aux méthodes (classe des protéines ayant une faible résolution, classe des protéines artificielles...) ; 2. fold : la composition en structures secondaires (hélices a et feuillets b), leur arrangement spatial et leurs connexions sont similaires ; 3. superfamily : l identité de séquence peut être faible mais où les structures et les fonctions suggèrent une origine évolutive commune ; 4. family : les structures protéiques ont au moins 30% d identité de séquence, ou bien possèdent des fonctions et des structures très similaires. La banque SCOP est donc une classification «manuelle» de domaines, et la définition des domaines est évidemment critique pour cette classification. La classification SCOP contenait en octobre 2004 pour entrées (70859 domaines): 945 classes au niveau fold et 1539 classes au niveau superfamily (2845 au niveau "families"). SCOP: Structural Classification of Proteins release, PDB Entries (1 Oct 2004) Domains (excluding nucleic acids and theoretical models) Class Number of folds Number of superfamilies Number of families All alpha proteins All beta proteins
.")
3 α and β proteins (α/β) α and β proteins (α+β) Multi-domain proteins Membrane and cell surface proteins Small proteins Total Classification CATH La classification CATH (Pearl et al., 2005; Pearl et al., 2000; Orengo et al., 1997) est effectuée à la fois automatiquement et manuellement. Comme SCOP, elle est hiérarchique et subdivisée en quatre niveaux principaux. Elle possède trois niveaux supplémentaires de classification établis sur la similarité des séquences protéiques. Les niveaux de classification sont : 1. Class où les structures sont regroupées selon leur composition en structures secondaires et les contacts entre celles-ci. Il y a quatre classes : mainly α et mainly β qui sont similaires aux deux classes all α et all β de SCOP, mixed α/β et Few secondary structures. L assignation d une structure à l une de ces quatre classes est automatique dans 90% des cas (les 10% restant sont assignés à la main) (Michie et al., 1996) ; 2. Architecture où l organisation générale des structures secondaires est la même pour les structures d un même groupe. Cette classification est faite manuellement, et notamment par rapport à la classification de J. Richardson (Richardson, 1981) ; 3. Topology : où les structures ayant un même repliement en terme de nombre, ordre et connexions de structures secondaires sont regroupées. La méthode de comparaison de deux structures SSAP (Taylor and Orengo, 1989b) est utilisée, avec une contrainte sur la longueur de l alignement et le score obtenu. ; 4. Homologous surperfamiliy où les structures d un même groupe ont des structures et des fonctions très similaires, suggérant un ancêtre commun. SSAP est aussi utilisé ; 5. Les niveaux supplémentaires - et imbriqués - S,N,I regroupent les structures ayant une identité de séquence respectivement > 35%, >95% et de 100% (ce dernier niveau regroupe en fait les protéines qui ont été résolues plusieurs fois, par exemple complexées ou non avec leur ligand). L algorithme d alignement des séquences est celui de Needleman et Wunsch. Les structures sont découpées en domaines structuraux selon le consensus trouvé par trois méthodes indépendantes, DETECTIVE (Swindells, 1995), PUU (Holm and Sander, 1994b) et DOMAK (Siddiqui and Barton, 1995). Si les trois méthodes s accordent pour le nombre de domaines et si 85% des résidus d un domaine sont les mêmes, le découpage de DETECTIVE est choisi, sinon, le découpage est fait à la main (47% des cas). Les structures ayant plus de 30 résidus hors domaines sont aussi découpées à la main. Le découpage en domaines structuraux n est effectué que pour une structure représentative de groupe du niveau N, les protéines du même groupe ayant plus de 95% d identité héritent des mêmes domaines. La cohérence du découpage en domaines est vérifiée au niveau supérieur S. Le protocole de classification a un peu varié au cours des années (Pearl et al., 2001). Pour ajouter une nouvelle structure à la classification, cette structure est comparée à la fois au niveau de sa séquence (alignement contre les séquences représentatives et contre des profils PSSM de PSIBLAST) et de sa structure (au niveau peptidique avec SSAP et au niveau des structures secondaires avec GRATH (Harrison et al., 2003)). Des alignements multiples des structures sont réalisés avec la méthode CORA (Orengo, 1999). L assignation automatique à une classe (Michie et al., 1996) commence par la détermination des éléments de structure secondaire par SSTRUC qui est une implémentation locale de DSSP

4 (Kabsch and Sander, 1983). Les structures secondaires sont ensuite représentées par des vecteurs et les distances internes entre Ca de deux structures secondaires sont calculés. Le nombre de structures secondaires et les contacts entre structures permet d établir la classification de la structure dans l une des quatre classes. En 2005, CATH contenait 1467 familles au niveau H dont 334 possédaient au moins 3 structures avec moins de 35% d identité (Pearl et al., 2005) et 813 classes au niveau T. CATH est donc une classification semi-automatique des structures. Familles de structures (résumé) On peut estimer selon les différentes classifications qu'il y a environ 1500 familles de structures. La distribution du nombre de protéines par familles semble suivre une loi de puissance (y=x -a ). Dans une étude menée de façon automatique (Carpentier, 2005, thèse), la famille la plus peuplée contient 223 structures et seules 4 familles sont constituées de plus de 100 protéines. La taille moyenne des familles est de 6,6 structures, et 50% des familles contiennent 3 structures ou moins. En considèrant les protéines comme des points dans un espace à haute dimension - espace des «formes» où les distances structurales entre protéines sont respectées-, Holm et Sander (Holm and Sander, 1996c) ont mis en évidence cinq «attracteurs» pour toutes les structures (voir figure ci dessous). Ces attracteurs servent à classer les domaines de protéines selon leur structure, l attribution d un domaine à un attracteur se faisant par un critère de chemin le plus court (deux classes s ajoutent, l une pour les structures non «connectées» aux autres, et l une pour les domaines à égale distance de deux attracteurs). 40% de tous les domaines connus sont couverts par 16 classes de repliements (figure A). Plusieurs paramètres raffinent cette première classification structurale : les alignements de séquence de PSIBLAST, l analyse des regroupements de résidus conservés, la classe enzymatique (EC number) et une analyse par mots clés de la fonction biologique.
 On peut estimer selon les différentes classifications qu'il y a environ 1500 familles de structures.")
5 A) La quantification des similarités des paires de structures (comparaison «~tout contre tout~») donne la position d'une structure dans un espace abstrait de hautes dimensions. La hauteur des pics reflète la densité de population de repliements, les axes horizontaux sont les axes des deux premiers vecteurs propres (i.e. associés aux deux plus grandes valeurs propres), l'axe vertical donne le nombre de repliements. La distribution des architectures est donnée par la projection sur le plan (la proximité sur ce plan donne une indication sur la similarité structurale entre 2 protéines) B) 40% de tous les domaines connus sont couverts par 16 classes de repliements. Ces 16 repliements sont montrés ici sous forme de diagrammes topologiques de structures secondaires dans la classe de leur attracteur (le numéro d'attracteur est le même que dans la figure A). Figures tirées de Holm et Sander (1996) "Mapping the protein universe" Méthodes de prédiction de structures Trois grands types de méthodes sont utilisées pour la prédiction de la structure 3D des protéines : la modélisation par «homologie», la reconnaissance de repliements., les méthodes de prédiction ab initio. La modélisation par «homologie» La modélisation par «homologie» s appuie sur l hypothèse que des protéines homologues ont en général conservé la même fonction et une topologie et une structure proches. Pour établir l homologie entre une séquence et une structure connue, il faut qu elles présentent une

6 similarité suffisante au niveau de leur séquence en acides aminés après alignement. On considère généralement qu'une identité de 30 à 35% suffit pour établir une modélisation correcte par homologie. Pour des séquences à >50% d'identité les modèles dérivés sont d une assez grande précision et justesse : ~ 1 angstrom de rms pour les atomes de la chaîne principale. Les erreurs se trouvent dans l empaquetage et l orientation des chaînes latérales. Cette précision suffit pour la prédiction de protéines partenaires ou le docking. Entre 30-50% d'identité de séquence les modèles sont d une précision et d une justesse moyenne. En plus des orientations, il y a des distortions dans le coeur, des erreurs de modélisation des boucles et des erreurs d alignement occasionnelles. Les rms sont typiquement ~1.5 angstrom. La modélisation peut servir dans ce cas dans la méthode de remplacement moléculaire en cristallographie, la détermination des épitopes, éventuellement pour du screening in silico de petits ligands. En dessous de 30% d'identité, les erreurs d alignement augmentent énormément. Ces erreurs sont les plus graves car elles peuvent résulter en un modèle de repliement totalement incorrect. Il faut utiliser des techniques ab-initio ou de reconnaissance de repliement. En ce cas, on ne peut que chercher à prédire des motifs fonctionnels, résidus conservés, ou en cristallographie utiliser ces structures pour l'ajustement dans des cartes électronique basse résolution. La modélisation ab initio / de novo Les méthodes ab initio sont fondées sur le paradigme que l état natif d une protéine est à son minimum global d énergie libre et cherchent à mener une recherche de ce minimum par un parcours à grande échelle de l espace conformationel. La clef de ces méthodes est d avoir une fonction «énergie» adéquate et une procédure efficace pour parcourir l espace des conformations. Pour cette raison, ces méthodes utilisent souvent un sous-ensemble des atomes de la chaîne peptidique et des chaînes latérales. La fonction énergie employée doit donc refléter les atomes omis et le solvant. Une des méthodes les plus connues et efficace, Rosetta (Simons et al., 1999), fait varier la structure de petits segments (3 et 9) entre des conformations compatibles avec la séquence locale mais ayant des conformations différentes (ces fragments sont précalculés sur les protéines connues de la PDB). Un grand nombre de conformations globales sont générées par combinaison de ces segments locaux (ce qui échantillonne l espace conformationel) et la fonction énergie est évaluée sur ces structures. Les prédictions de novo, à l aide d alphabets structuraux (de Brevern et al., 2001) sont à classer parmi ces méthodes qui ne s appuient pas sur une homologie particulière avec une structure mais plutôt sur un apprentissage de ce qu «est» une structure (petits blocs chevauchants, corrélation de l enchaînement de ces blocs). L annotation structurale- génomique structurale Le séquençage systématique des génomes demande le développement de méthodes d annotation nouvelles afin de mieux reconnaître les gènes ayant divergé. Les méthodes usuelles de l annotation s appuient sur les biais statistiques liés aux gènes (biais de codage, biais d usage du code), sur la reconnaissance de signaux génétiques (start, stop, promoteurs, sites de fixation du ribosome, etc), et surtout sur la similarité des phases ouvertes de lecture avec des séquences déjà caractérisées. Les outils de détection de similarités de séquence généralement utilisés pour établir cette similarité sont BLAST (Altschul et al., 1990; Altschul et al., 1997) et FASTA (Pearson and Lipman, 1988; Pearson, 2000). La dégénérescence bien connue entre séquences et structures (Chothia and Gerstein, 1997) limite ces méthodes puisqu il sera difficile de détecter la similarité entre séquences ayant divergé depuis longtemps. Ainsi, réglés à un seuil de détection de 1% de faux positifs, BLAST ou FASTA ne

7 reconnaissent comme significatives que 15% de séquences apparentées et possédant moins de 40% de résidus conservés entre elles (Brenner et al., 1998). La détection de similarités est améliorée par des méthodes basées sur des alignements mutliples (Park et al., 1998), ou «profils», comme PSIBLAST (Altschul et al., 1997). Pour aller plus loin dans la détection de similarités, les méthodes de reconnaissance de repliements sont des outils de grand intérêt. Elles permettent de détecter des similarités indécelables grâce aux seules séquences. De plus, des programmes de résolution de structures 3D à grande échelle sont lancés aujourd'hui. Pour être efficace, il ne faut pas chercher à résoudre la structure d'une protéine homologue à une structure déjà connue. Donc, on cherche des séquences qui ne ressemblent à aucune protéine dont la structure 3D serait connue, avec des techniques d'analyse de séquence, comme PSIBLAST, mais aussi avec des techniques de reconnaissance de repliement plus fines. La reconnaissance de repliements ("Threading") Les méthodes de reconnaissance de repliements ou de "threading" reposent sur l observation que les structures 3D sont mieux conservées que les séquences (Chothia and Lesk, 1986) et qu ainsi les structures constituent un meilleur support pour caractériser des protéines homologues distantes que leur séquences. De plus, le nombre de repliements différents estimé est de plusieurs ordres de grandeur en dessous du nombre de familles de séquences différentes (dégénérescence structure-séquence). Ce nombre est estimé de 1000 à 8000 selon les études (Chothia, 1992; Orengo et al., 1994; Wang, 1996; Wang, 1998; Govindarajan et al., 1999). Les méthodes de threading (Smith et al., 1997) visent donc à calculer l adéquation entre une séquence de structure inconnue et une structure 3D connue (ou parties de celle-ci). La séquence est «enfilée» sur chaque repliement d une librairie de coeurs représentatifs de la PDB. La compatibilité de la séquence avec un repliement particulier peut se mesurer grâce à un potentiel empirique : potentiels statistiques de distances entre résidus, précalculés sur la PDB (Sippl, 1990) ou scores de paires de résidus étant donné la structure secondaire où se trouvent ceux-ci. Les techniques de "threading" ("montage") sont basées sur la méthode suivante: 1) une librairie de repliements protéiques non redondants ("coeurs") est constituée à partir de la banque de données structurale des protéines (PDB Brookhaven). Chaque repliement est une chaîne tridimensionnelle, la séquence étant complètement oubliée. 2) La séquence test est ensuite ajustée de manière optimale à chaque repliement de la librairie (insertions-délétions permises dans les boucles), l' "énergie" de chaque ajustement ("montage") étant calculé en sommant les "interactions" des résidus deux à deux. Les repliements de la librairie sont ensuite rangés par ordre d'énergie, le plus probable pour la séquence étant celui d'énergie la plus basse. Potentiels utilisés en threading Dans les premiers travaux, le problème difficile - optimiser le "montage" de la séquence test sur une structure d'après les interactions deux à deux - était évité en ajustant en fait sur la séquence primaire du "coeur" (le niveau le plus bas se ramenant à un alignement de la séquence du coeur avec la séquence test, en calculant un score résidus à résidus dérivé d'une matrice de similarité genre Dayhoff (PAM250)). Vu la dégénerescence séquence-structure (1 structure pouvant correspondre à plusieurs séquences présentant une faible similarité), il était plus tentant de décrire le "coeur" non pas en termes de séquence primaire, mais en terme d'environnement de chaque position dans la structure. En effet, l'environnement - par exemple la structure secondaire locale, accessibilité au solvant,... - d'une position tend à être plus conservé que la nature du résidu à cette position,

8 et une méthode qui tient compte de cet environnement sera plus à même de détecter des relations séquence-structure qu'une méthode basée uniquement sur la séquence. Pour évaluer l'énergie d'une séquence dans une conformation particulière (un "montage" particulier), on a besoin d'un ensemble de potentiels pour l'interaction entre les résidus, qui n'ait pas besoin d'une modélisation explicite des chaînes latérales (on a montré que les potentiels de mécanique moléculaire classiques ne peuvent discerner si une protéine est dans son repliement natif ou non). On utilise donc des potentiels basés sur des statistiques d'observables sur les structures connues (par exemple: la distance entre deux résidus de natures données), et on prend en compte explicitement le degré de solvatation des résidus. Pour une paire d'amino-acides donnés, une séparation topologique (distance sur la séquence) et une distance d'interaction (distance dans l'espace tridimensionnel), ces potentiels permettent la mesure d'une pseudo-énergie relative à la probabilité d'observer dans les protéines natives les interactions effectivement observées pour le "montage" donné. Par exemple, pour les deux Cβ d'une paire de résidus a et b, à une séparation topologique (sur la séquence) de k, et à un intervalle de distance physique de s, le potentiel est donné par (Sippl, 1990, J. Mol. Biol.,1993) : E k ab = RT ln[1+m ab σ] - RT ln[1+ m ab σ Erreur!], où m ab est le nombre de paires ab observées à distance k, σ est le poids donné à chaque observation, f k (s) est la fréquence de toutes les paires de résidus à une distance topologique k et une distance physique s, et f k ab (s) est la fréquence équivalente pour la paire ab. Les potentiels sont calculés pour les paires Cβ Cβ, Cβ N, Cβ O, N Cβ, N O, O Cβ et O N. De même, le potentiel pour la solvatation d'un acide aminé a est défini par: E solv a (r) = -RT ln[ Erreur!], où r est le pourcentage d'accessibilité (relatif à une accessibilité dans GGXGG étendu), f a (r) est la fréquence d'un résidu a avec une accessibilité r, et f(r) est la fréquence de tous les résidus avec une accessibilité r (calculée avec le programme DSSP par exemple). Ces potentiels sont de type Boltzman, la fonction de partition des conformations est du type Z = e - G/kT (Z=nombre d'une conformation donnée/total des conformations). En séparant ces potentiels empiriques par intervalles de portée, on peut tenter d'évaluer une signification structurale spécifique pour chaque intervalle d'interaction. Par exemple, les termes à courte portée prédominent dans l'ajustement des éléments de structure secondaire: en "montant" une séquence sur une hélice α et en évaluant ces termes potentiels à courte portée, la probabilité que la séquence se replie en hélice α peut être calculée. De même, les interactions intermédiaires règlent les motifs super-secondaires, et les interactions longue distance, la structure tertiaire. Les différents auteurs ne prennent pas les mêmes critères pour les interactions. Par exemple, on peut prendre en compte aussi la nature des acides aminés de la séquence du "coeur" dans le potentiel. A la place des interactions longues distances (> 10Å), Jones et coll. (Nature, 358, 1992 pp86-89) utilisent un "potentiel de solvatation" (qui est d'ailleurs la seule énergie évaluée pour les boucles)
, et on prend en compte")
9 a) Interactions à courte portée sur la séquence (distance topologique, k=3) Cβ-Cβ pour Ala- Ala. Les deux puits de potentiels observés correspondent principalement aux structures α (6 Å) et β (9 Å). b) Interactions à longues portée (distance tridimensionnelle) pour Cys-Cys Cβ-Cβ. Le puit de potentiel observé à 4 Å correspond aux ponts disulfure. c) Potentiel versus le pourcentage d'accessibilité au solvant pour la leucine d) Potentiel versus le pourcentage d'accessibilité au solvant pour l'acide glutamique. (tiré de Jones et coll. Nature,358,1992) Problème de la définition des "coeurs" pour les structures 3D: Il faut des repliements représentatifs : non redondance, qualité des structures Définir les régions conservées (coeur) : - structures secondaires + bornes inférieures et supérieures pour les boucles - bloc conservés en structure dans une famille (lorsqu'on a les structures!) 2) calcul de l'énergie empirique (sur les données disponibles dans les banques de structure): Erreur!= e(-ρ(r,s,dij p )) Potentiel de force moyenne ("mean force potential"), analogue à une fonction de répartition (voir Boltzman, Z = e - G/kT ).
 Problème de la définition des \"coeurs\" pour les structures 3D: Il faut des repliements représentatifs : non redondance, qualité des structures Définir les régions conservées (coeur)")
10 Le potentiel de force moyenne peut être constitué de plusieurs potentiels pondérés (leur nombre et leur choix variant selon les auteurs) : Méthode d'alignement de la séquence sur la structure: Selon les auteurs, la méthode d'alignement peut être: - Monte Carlo + recuit simulé (choix des segments, affinage (par calcul d'énergie): ajout ou suppression d'un résidu en bout de structure) - Programmation dynamique (comme en alignement de séquence) On peut transformer la représentation 3D en 1D: Coil, Helical, Extended, buried, exposed C C H H C C C E E C C C C e e e b b e b b b b e e e Ce Ce He Hb Cb Ce Cb Cb Eb Cb Ce Ce Ce M F T V N E H I D R L Y A Matrice de coût des interactions entre blocs.

11 Fonction évaluation 1D (calcul de scores de substitution basé sur des alignements multiples de familles de structures -> probabilités) Méthodes d'alignement: programmation dynamique avec des matrices scores-position (PSSM: position specific scoring matrix)

12
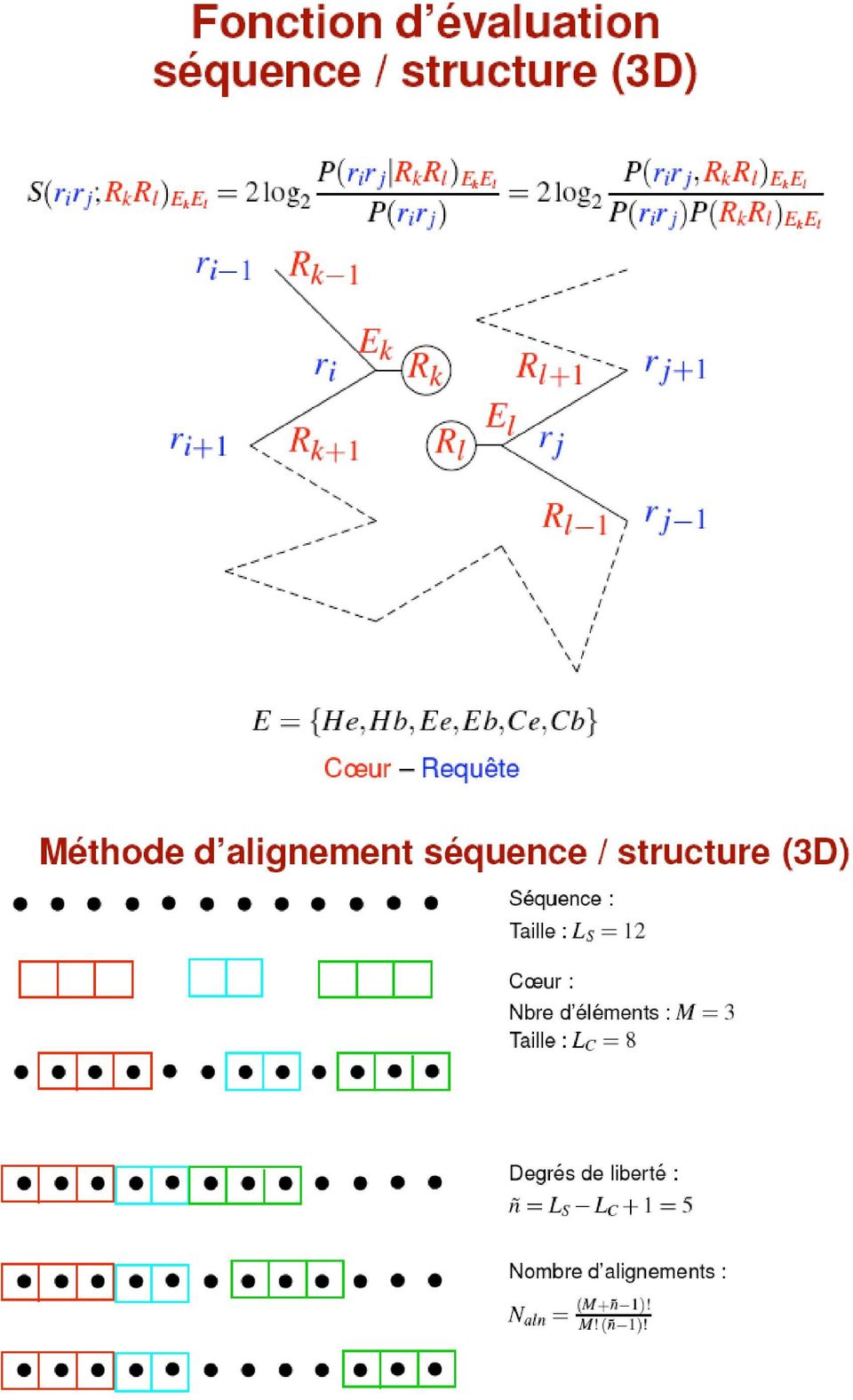
13 Statistique des scores: Il est impossible de comparer directement les scores obtenus par une même séquence sur différents coeurs. - il faut donc une normalisation des scores: modèle statistique ou expérimental

14 JOMPO A FINIR 1) distribution d'énergie pour permutations aléatoires de l'alignement (dans les zones alignées) -> z-score z 0 = <E>-E0/σ pour le "montage" sur un coeur). Cela ne suffit pas-> 2) calcul de z avec des séquences aléatoires de même composition.
 calcul de z avec des séquences")
Formavie 2010. 2 Différentes versions du format PDB...3. 3 Les champs dans les fichiers PDB...4. 4 Le champ «ATOM»...5. 6 Limites du format PDB...
 Formavie 2010 Les fichiers PDB Les fichiers PDB contiennent les informations qui vont permettre à des logiciels de visualisation moléculaire (ex : RasTop ou Jmol) d afficher les molécules. Un fichier au
Formavie 2010 Les fichiers PDB Les fichiers PDB contiennent les informations qui vont permettre à des logiciels de visualisation moléculaire (ex : RasTop ou Jmol) d afficher les molécules. Un fichier au
IMMUNOLOGIE. La spécificité des immunoglobulines et des récepteurs T. Informations scientifiques
 IMMUNOLOGIE La spécificité des immunoglobulines et des récepteurs T Informations scientifiques L infection par le VIH entraîne des réactions immunitaires de l organisme qui se traduisent par la production
IMMUNOLOGIE La spécificité des immunoglobulines et des récepteurs T Informations scientifiques L infection par le VIH entraîne des réactions immunitaires de l organisme qui se traduisent par la production
Identification de nouveaux membres dans des familles d'interleukines
 Identification de nouveaux membres dans des familles d'interleukines Nicolas Beaume Jérôme Mickolajczak Gérard Ramstein Yannick Jacques 1ère partie : Définition de la problématique Les familles de gènes
Identification de nouveaux membres dans des familles d'interleukines Nicolas Beaume Jérôme Mickolajczak Gérard Ramstein Yannick Jacques 1ère partie : Définition de la problématique Les familles de gènes
La reconnaissance moléculaire: la base du design rationnel Modélisation moléculaire: Introduction Hiver 2006
 La reconnaissance moléculaire: la base du design rationnel En 1890 Emil Fisher a proposé le modèle "serrure et clé" pour expliquer la façon de fonctionner des systèmes biologiques. Un substrat rentre et
La reconnaissance moléculaire: la base du design rationnel En 1890 Emil Fisher a proposé le modèle "serrure et clé" pour expliquer la façon de fonctionner des systèmes biologiques. Un substrat rentre et
INF6304 Interfaces Intelligentes
 INF6304 Interfaces Intelligentes filtres collaboratifs 1/42 INF6304 Interfaces Intelligentes Systèmes de recommandations, Approches filtres collaboratifs Michel C. Desmarais Génie informatique et génie
INF6304 Interfaces Intelligentes filtres collaboratifs 1/42 INF6304 Interfaces Intelligentes Systèmes de recommandations, Approches filtres collaboratifs Michel C. Desmarais Génie informatique et génie
Big data et sciences du Vivant L'exemple du séquençage haut débit
 Big data et sciences du Vivant L'exemple du séquençage haut débit C. Gaspin, C. Hoede, C. Klopp, D. Laborie, J. Mariette, C. Noirot, MS. Trotard [email protected] INRA - MIAT - Plate-forme
Big data et sciences du Vivant L'exemple du séquençage haut débit C. Gaspin, C. Hoede, C. Klopp, D. Laborie, J. Mariette, C. Noirot, MS. Trotard [email protected] INRA - MIAT - Plate-forme
Optimisation de la compression fractale D images basée sur les réseaux de neurones
 Optimisation de la compression fractale D images basée sur les réseaux de neurones D r BOUKELIF Aoued Communication Networks,Architectures and Mutimedia laboratory University of S.B.A [email protected]
Optimisation de la compression fractale D images basée sur les réseaux de neurones D r BOUKELIF Aoued Communication Networks,Architectures and Mutimedia laboratory University of S.B.A [email protected]
Prédiction de la structure d une
 Prédiction de la structure d une protéine Soluscience Guillaume Chakroun guillaume [email protected] Copyright c 2004 Guillaume Chakroun TABLE DES MATIÈRES Table des matières 1 Les structures protéiques
Prédiction de la structure d une protéine Soluscience Guillaume Chakroun guillaume [email protected] Copyright c 2004 Guillaume Chakroun TABLE DES MATIÈRES Table des matières 1 Les structures protéiques
MABioVis. Bio-informatique et la
 MABioVis Modèles et Algorithmes pour la Bio-informatique et la Visualisation Visite ENS Cachan 5 janvier 2011 MABioVis G GUY MELANÇON (PR UFR Maths Info / EPI GRAVITE) (là, maintenant) - MABioVis DAVID
MABioVis Modèles et Algorithmes pour la Bio-informatique et la Visualisation Visite ENS Cachan 5 janvier 2011 MABioVis G GUY MELANÇON (PR UFR Maths Info / EPI GRAVITE) (là, maintenant) - MABioVis DAVID
Sauvegarde collaborative entre pairs Ludovic Courtès LAAS-CNRS
 Sauvegarde collaborative entre pairs 1 Sauvegarde collaborative entre pairs Ludovic Courtès LAAS-CNRS Sauvegarde collaborative entre pairs 2 Introduction Pourquoi pair à pair? Utilisation de ressources
Sauvegarde collaborative entre pairs 1 Sauvegarde collaborative entre pairs Ludovic Courtès LAAS-CNRS Sauvegarde collaborative entre pairs 2 Introduction Pourquoi pair à pair? Utilisation de ressources
INTRODUCTION À L'ENZYMOLOGIE
 INTRODUCTION À L'ENZYMOLOGIE Les enzymes sont des macromolécules spécialisées qui - catalysent les réactions biologiques - transforment différentes formes d'énergie. Les enzymes diffèrent des catalyseurs
INTRODUCTION À L'ENZYMOLOGIE Les enzymes sont des macromolécules spécialisées qui - catalysent les réactions biologiques - transforment différentes formes d'énergie. Les enzymes diffèrent des catalyseurs
Annexe commune aux séries ES, L et S : boîtes et quantiles
 Annexe commune aux séries ES, L et S : boîtes et quantiles Quantiles En statistique, pour toute série numérique de données à valeurs dans un intervalle I, on définit la fonction quantile Q, de [,1] dans
Annexe commune aux séries ES, L et S : boîtes et quantiles Quantiles En statistique, pour toute série numérique de données à valeurs dans un intervalle I, on définit la fonction quantile Q, de [,1] dans
Rapport d'analyse des besoins
 Projet ANR 2011 - BR4CP (Business Recommendation for Configurable products) Rapport d'analyse des besoins Janvier 2013 Rapport IRIT/RR--2013-17 FR Redacteur : 0. Lhomme Introduction...4 La configuration
Projet ANR 2011 - BR4CP (Business Recommendation for Configurable products) Rapport d'analyse des besoins Janvier 2013 Rapport IRIT/RR--2013-17 FR Redacteur : 0. Lhomme Introduction...4 La configuration
La gestion de données dans le cadre d une application de recherche d alignement de séquence : BLAST.
 La gestion de données dans le cadre d une application de recherche d alignement de séquence : BLAST. Gaël Le Mahec - p. 1/12 L algorithme BLAST. Basic Local Alignment Search Tool est un algorithme de recherche
La gestion de données dans le cadre d une application de recherche d alignement de séquence : BLAST. Gaël Le Mahec - p. 1/12 L algorithme BLAST. Basic Local Alignment Search Tool est un algorithme de recherche
Transmission d informations sur le réseau électrique
 Transmission d informations sur le réseau électrique Introduction Remarques Toutes les questions en italique devront être préparées par écrit avant la séance du TP. Les préparations seront ramassées en
Transmission d informations sur le réseau électrique Introduction Remarques Toutes les questions en italique devront être préparées par écrit avant la séance du TP. Les préparations seront ramassées en
CHAPITRE 3 LA SYNTHESE DES PROTEINES
 CHAITRE 3 LA SYNTHESE DES ROTEINES On sait qu un gène détient dans sa séquence nucléotidique, l information permettant la synthèse d un polypeptide. Ce dernier caractérisé par sa séquence d acides aminés
CHAITRE 3 LA SYNTHESE DES ROTEINES On sait qu un gène détient dans sa séquence nucléotidique, l information permettant la synthèse d un polypeptide. Ce dernier caractérisé par sa séquence d acides aminés
Développement itératif, évolutif et agile
 Document Développement itératif, évolutif et agile Auteur Nicoleta SERGI Version 1.0 Date de sortie 23/11/2007 1. Processus Unifié Développement itératif, évolutif et agile Contrairement au cycle de vie
Document Développement itératif, évolutif et agile Auteur Nicoleta SERGI Version 1.0 Date de sortie 23/11/2007 1. Processus Unifié Développement itératif, évolutif et agile Contrairement au cycle de vie
LA NOTATION STATISTIQUE DES EMPRUNTEURS OU «SCORING»
 LA NOTATION STATISTIQUE DES EMPRUNTEURS OU «SCORING» Gilbert Saporta Professeur de Statistique Appliquée Conservatoire National des Arts et Métiers Dans leur quasi totalité, les banques et organismes financiers
LA NOTATION STATISTIQUE DES EMPRUNTEURS OU «SCORING» Gilbert Saporta Professeur de Statistique Appliquée Conservatoire National des Arts et Métiers Dans leur quasi totalité, les banques et organismes financiers
Vision industrielle et télédétection - Détection d ellipses. Guillaume Martinez 17 décembre 2007
 Vision industrielle et télédétection - Détection d ellipses Guillaume Martinez 17 décembre 2007 1 Table des matières 1 Le projet 3 1.1 Objectif................................ 3 1.2 Les choix techniques.........................
Vision industrielle et télédétection - Détection d ellipses Guillaume Martinez 17 décembre 2007 1 Table des matières 1 Le projet 3 1.1 Objectif................................ 3 1.2 Les choix techniques.........................
Objectifs. Clustering. Principe. Applications. Applications. Cartes de crédits. Remarques. Biologie, Génomique
 Objectifs Clustering On ne sait pas ce qu on veut trouver : on laisse l algorithme nous proposer un modèle. On pense qu il existe des similarités entre les exemples. Qui se ressemble s assemble p. /55
Objectifs Clustering On ne sait pas ce qu on veut trouver : on laisse l algorithme nous proposer un modèle. On pense qu il existe des similarités entre les exemples. Qui se ressemble s assemble p. /55
Déroulement d un projet en DATA MINING, préparation et analyse des données. Walid AYADI
 1 Déroulement d un projet en DATA MINING, préparation et analyse des données Walid AYADI 2 Les étapes d un projet Choix du sujet - Définition des objectifs Inventaire des données existantes Collecte, nettoyage
1 Déroulement d un projet en DATA MINING, préparation et analyse des données Walid AYADI 2 Les étapes d un projet Choix du sujet - Définition des objectifs Inventaire des données existantes Collecte, nettoyage
données en connaissance et en actions?
 1 Partie 2 : Présentation de la plateforme SPSS Modeler : Comment transformer vos données en connaissance et en actions? SPSS Modeler : l atelier de data mining Large gamme de techniques d analyse (algorithmes)
1 Partie 2 : Présentation de la plateforme SPSS Modeler : Comment transformer vos données en connaissance et en actions? SPSS Modeler : l atelier de data mining Large gamme de techniques d analyse (algorithmes)
Conception de Médicament
 Conception de Médicament Approche classique HTS Chimie combinatoire Rational Drug Design Ligand based (QSAR) Structure based (ligand et ou macromolec.) 3DQSAR Docking Virtual screening Needle in a Haystack
Conception de Médicament Approche classique HTS Chimie combinatoire Rational Drug Design Ligand based (QSAR) Structure based (ligand et ou macromolec.) 3DQSAR Docking Virtual screening Needle in a Haystack
EXPLOITATIONS PEDAGOGIQUES DU TABLEUR EN STG
 Exploitations pédagogiques du tableur en STG Académie de Créteil 2006 1 EXPLOITATIONS PEDAGOGIQUES DU TABLEUR EN STG Commission inter-irem lycées techniques contact : [email protected] La maquette
Exploitations pédagogiques du tableur en STG Académie de Créteil 2006 1 EXPLOITATIONS PEDAGOGIQUES DU TABLEUR EN STG Commission inter-irem lycées techniques contact : [email protected] La maquette
Cloud Computing et SaaS
 Cloud Computing et SaaS On a vu fleurir ces derniers temps un grands nombre de sigles. L un des premiers est SaaS, Software as a Service, sur lequel nous aurons l occasion de revenir. Mais il y en a beaucoup
Cloud Computing et SaaS On a vu fleurir ces derniers temps un grands nombre de sigles. L un des premiers est SaaS, Software as a Service, sur lequel nous aurons l occasion de revenir. Mais il y en a beaucoup
Perl Orienté Objet BioPerl There is more than one way to do it
 Perl Orienté Objet BioPerl There is more than one way to do it Bérénice Batut, [email protected] DUT Génie Biologique Option Bioinformatique Année 2014-2015 Perl Orienté Objet - BioPerl Rappels
Perl Orienté Objet BioPerl There is more than one way to do it Bérénice Batut, [email protected] DUT Génie Biologique Option Bioinformatique Année 2014-2015 Perl Orienté Objet - BioPerl Rappels
ProxiLens : Exploration interactive de données multidimensionnelles à partir de leur projection
 ProxiLens : Exploration interactive de données multidimensionnelles à partir de leur projection Nicolas HEULOT (CEA LIST) Michaël AUPETIT (CEA LIST) Jean-Daniel FEKETE (INRIA Saclay) Journées Big Data
ProxiLens : Exploration interactive de données multidimensionnelles à partir de leur projection Nicolas HEULOT (CEA LIST) Michaël AUPETIT (CEA LIST) Jean-Daniel FEKETE (INRIA Saclay) Journées Big Data
Baccalauréat L spécialité, Métropole et Réunion, 19 juin 2009 Corrigé.
 Baccalauréat L spécialité, Métropole et Réunion, 19 juin 2009 Corrigé. L usage d une calculatrice est autorisé Durée : 3heures Deux annexes sont à rendre avec la copie. Exercice 1 5 points 1_ Soit f la
Baccalauréat L spécialité, Métropole et Réunion, 19 juin 2009 Corrigé. L usage d une calculatrice est autorisé Durée : 3heures Deux annexes sont à rendre avec la copie. Exercice 1 5 points 1_ Soit f la
Limitations of the Playstation 3 for High Performance Cluster Computing
 Introduction Plan Limitations of the Playstation 3 for High Performance Cluster Computing July 2007 Introduction Plan Introduction Intérêts de la PS3 : rapide et puissante bon marché L utiliser pour faire
Introduction Plan Limitations of the Playstation 3 for High Performance Cluster Computing July 2007 Introduction Plan Introduction Intérêts de la PS3 : rapide et puissante bon marché L utiliser pour faire
Chapitre 1. L intérêt. 2. Concept d intérêt. 1. Mise en situation. Au terme de ce chapitre, vous serez en mesure de :
 Chapitre 1 L intérêt Au terme de ce chapitre, vous serez en mesure de : 1. Comprendre la notion générale d intérêt. 2. Distinguer la capitalisation à intérêt simple et à intérêt composé. 3. Calculer la
Chapitre 1 L intérêt Au terme de ce chapitre, vous serez en mesure de : 1. Comprendre la notion générale d intérêt. 2. Distinguer la capitalisation à intérêt simple et à intérêt composé. 3. Calculer la
Relation entre deux variables : estimation de la corrélation linéaire
 CHAPITRE 3 Relation entre deux variables : estimation de la corrélation linéaire Parmi les analyses statistiques descriptives, l une d entre elles est particulièrement utilisée pour mettre en évidence
CHAPITRE 3 Relation entre deux variables : estimation de la corrélation linéaire Parmi les analyses statistiques descriptives, l une d entre elles est particulièrement utilisée pour mettre en évidence
Semestre 2 Spécialité «Analyse in silico des complexes macromolécules biologiques-médicaments»
 Master In silico Drug Design Semestre 2 Spécialité «Analyse in silico des complexes macromolécules biologiques-médicaments» 30NU01IS INITIATION A LA PROGRAMMATION (6 ECTS) Responsables : D. MESTIVIER,
Master In silico Drug Design Semestre 2 Spécialité «Analyse in silico des complexes macromolécules biologiques-médicaments» 30NU01IS INITIATION A LA PROGRAMMATION (6 ECTS) Responsables : D. MESTIVIER,
Infolettre #18 : Les graphiques avec Excel 2010
 Infolettre #18 : Les graphiques avec Excel 2010 Table des matières Introduction... 1 Hourra! Le retour du double-clic... 1 Modifier le graphique... 4 Onglet Création... 4 L onglet Disposition... 7 Onglet
Infolettre #18 : Les graphiques avec Excel 2010 Table des matières Introduction... 1 Hourra! Le retour du double-clic... 1 Modifier le graphique... 4 Onglet Création... 4 L onglet Disposition... 7 Onglet
Cours n 12. Technologies WAN 2nd partie
 Cours n 12 Technologies WAN 2nd partie 1 Sommaire Aperçu des technologies WAN Technologies WAN Conception d un WAN 2 Lignes Louées Lorsque des connexions dédiées permanentes sont nécessaires, des lignes
Cours n 12 Technologies WAN 2nd partie 1 Sommaire Aperçu des technologies WAN Technologies WAN Conception d un WAN 2 Lignes Louées Lorsque des connexions dédiées permanentes sont nécessaires, des lignes
Partie 7 : Gestion de la mémoire
 INF3600+INF2610 Automne 2006 Partie 7 : Gestion de la mémoire Exercice 1 : Considérez un système disposant de 16 MO de mémoire physique réservée aux processus utilisateur. La mémoire est composée de cases
INF3600+INF2610 Automne 2006 Partie 7 : Gestion de la mémoire Exercice 1 : Considérez un système disposant de 16 MO de mémoire physique réservée aux processus utilisateur. La mémoire est composée de cases
Critères pour les méthodes de quantification des résidus potentiellement allergéniques de protéines de collage dans le vin (OIV-Oeno 427-2010)
") Méthode OIV- -MA-AS315-23 Type de méthode : critères Critères pour les méthodes de quantification des résidus potentiellement allergéniques de protéines de collage (OIV-Oeno 427-2010) 1 Définitions des
Méthode OIV- -MA-AS315-23 Type de méthode : critères Critères pour les méthodes de quantification des résidus potentiellement allergéniques de protéines de collage (OIV-Oeno 427-2010) 1 Définitions des
Fonctions de plusieurs variables
 Module : Analyse 03 Chapitre 00 : Fonctions de plusieurs variables Généralités et Rappels des notions topologiques dans : Qu est- ce que?: Mathématiquement, n étant un entier non nul, on définit comme
Module : Analyse 03 Chapitre 00 : Fonctions de plusieurs variables Généralités et Rappels des notions topologiques dans : Qu est- ce que?: Mathématiquement, n étant un entier non nul, on définit comme
Projet SINF2275 «Data mining and decision making» Projet classification et credit scoring
 Projet SINF2275 «Data mining and decision making» Projet classification et credit scoring Année académique 2006-2007 Professeurs : Marco Saerens Adresse : Université catholique de Louvain Information Systems
Projet SINF2275 «Data mining and decision making» Projet classification et credit scoring Année académique 2006-2007 Professeurs : Marco Saerens Adresse : Université catholique de Louvain Information Systems
Baccalauréat ES Polynésie (spécialité) 10 septembre 2014 Corrigé
 10 septembre 2014 Corrigé") Baccalauréat ES Polynésie (spécialité) 10 septembre 2014 Corrigé A. P. M. E. P. Exercice 1 5 points 1. Réponse d. : 1 e Le coefficient directeur de la tangente est négatif et n est manifestement pas 2e
Baccalauréat ES Polynésie (spécialité) 10 septembre 2014 Corrigé A. P. M. E. P. Exercice 1 5 points 1. Réponse d. : 1 e Le coefficient directeur de la tangente est négatif et n est manifestement pas 2e
Confort, sécurité et gestion énergétique
 Fonctionnalités d une système de GTB Offre globale pour la productivité des bâtiments, Sécurité des personnes et des biens Sécurité Incendie Signalétique d évacuation Détection d Intrusion Vidéosurveillance
Fonctionnalités d une système de GTB Offre globale pour la productivité des bâtiments, Sécurité des personnes et des biens Sécurité Incendie Signalétique d évacuation Détection d Intrusion Vidéosurveillance
Travaux pratiques avec RapidMiner
 Travaux pratiques avec RapidMiner Master Informatique de Paris 6 Spécialité IAD Parcours EDOW Module Algorithmes pour la Fouille de Données Janvier 2012 Prise en main Généralités RapidMiner est un logiciel
Travaux pratiques avec RapidMiner Master Informatique de Paris 6 Spécialité IAD Parcours EDOW Module Algorithmes pour la Fouille de Données Janvier 2012 Prise en main Généralités RapidMiner est un logiciel
POLITIQUE SUR LA SÉCURITÉ LASER
 Date d entrée en vigueur: 30 aout 2011 Remplace/amende: VRS-51/s/o Origine: Vice-rectorat aux services Numéro de référence: VPS-51 Les utilisateurs de lasers devront suivre les directives, la politique
Date d entrée en vigueur: 30 aout 2011 Remplace/amende: VRS-51/s/o Origine: Vice-rectorat aux services Numéro de référence: VPS-51 Les utilisateurs de lasers devront suivre les directives, la politique
Mesure agnostique de la qualité des images.
 Mesure agnostique de la qualité des images. Application en biométrie Christophe Charrier Université de Caen Basse-Normandie GREYC, UMR CNRS 6072 Caen, France 8 avril, 2013 C. Charrier NR-IQA 1 / 34 Sommaire
Mesure agnostique de la qualité des images. Application en biométrie Christophe Charrier Université de Caen Basse-Normandie GREYC, UMR CNRS 6072 Caen, France 8 avril, 2013 C. Charrier NR-IQA 1 / 34 Sommaire
WEBINAIRE SUR LE SUIVI DE TENDANCES
 WEBINAIRE SUR LE SUIVI DE TENDANCES Le 16/02/2012 à 21H Présenté par Gilles SANTACREU (Boursikoter.com) En partenariat avec CMC Markets 1 Gilles SANTACREU, 43 ans - Webmaster et fondateur du site Boursikoter.com
WEBINAIRE SUR LE SUIVI DE TENDANCES Le 16/02/2012 à 21H Présenté par Gilles SANTACREU (Boursikoter.com) En partenariat avec CMC Markets 1 Gilles SANTACREU, 43 ans - Webmaster et fondateur du site Boursikoter.com
Sciences de Gestion Spécialité : SYSTÈMES D INFORMATION DE GESTION
 Sciences de Gestion Spécialité : SYSTÈMES D INFORMATION DE GESTION Classe de terminale de la série Sciences et Technologie du Management et de la Gestion Préambule Présentation Les technologies de l information
Sciences de Gestion Spécialité : SYSTÈMES D INFORMATION DE GESTION Classe de terminale de la série Sciences et Technologie du Management et de la Gestion Préambule Présentation Les technologies de l information
Annexe : La Programmation Informatique
 GLOSSAIRE Table des matières La Programmation...2 Les langages de programmation...2 Java...2 La programmation orientée objet...2 Classe et Objet...3 API et Bibliothèque Logicielle...3 Environnement de
GLOSSAIRE Table des matières La Programmation...2 Les langages de programmation...2 Java...2 La programmation orientée objet...2 Classe et Objet...3 API et Bibliothèque Logicielle...3 Environnement de
Industrie des cartes de paiement (PCI) Norme de sécurité des données Récapitulatif des modifications de
 Norme de sécurité des données Récapitulatif des modifications de") Industrie des cartes de paiement (PCI) Norme de sécurité des données Récapitulatif des modifications de la norme PCI DSS entre les versions 2.0 et 3.0 Novembre 2013 Introduction Ce document apporte un
Industrie des cartes de paiement (PCI) Norme de sécurité des données Récapitulatif des modifications de la norme PCI DSS entre les versions 2.0 et 3.0 Novembre 2013 Introduction Ce document apporte un
Jouve, 18, rue Saint-Denis, 75001 PARIS
 19 à Europâisches Patentamt European Patent Office Office européen des brevets Numéro de publication : 0 645 740 A1 12 DEMANDE DE BREVET EUROPEEN @ Numéro de dépôt : 94402079.1 @ Int. ci.6: G07B 17/04,
19 à Europâisches Patentamt European Patent Office Office européen des brevets Numéro de publication : 0 645 740 A1 12 DEMANDE DE BREVET EUROPEEN @ Numéro de dépôt : 94402079.1 @ Int. ci.6: G07B 17/04,
Systèmes de transmission
 Systèmes de transmission Conception d une transmission série FABRE Maxime 2012 Introduction La transmission de données désigne le transport de quelque sorte d'information que ce soit, d'un endroit à un
Systèmes de transmission Conception d une transmission série FABRE Maxime 2012 Introduction La transmission de données désigne le transport de quelque sorte d'information que ce soit, d'un endroit à un
Intelligence Artificielle et Systèmes Multi-Agents. Badr Benmammar [email protected]
 Intelligence Artificielle et Systèmes Multi-Agents Badr Benmammar [email protected] Plan La première partie : L intelligence artificielle (IA) Définition de l intelligence artificielle (IA) Domaines
Intelligence Artificielle et Systèmes Multi-Agents Badr Benmammar [email protected] Plan La première partie : L intelligence artificielle (IA) Définition de l intelligence artificielle (IA) Domaines
EBS 204 E C B S. Publication : Novembre 96
 EBS 204 E C B S Publication : Novembre 96 Traduction française de la norme internationale produite par le CENB en novembre 1996 0 INTRODUCTION 1 DOMAINE D'APPLICATION 2 REFERENCES NORMATIVES 3 DEFINITIONS
EBS 204 E C B S Publication : Novembre 96 Traduction française de la norme internationale produite par le CENB en novembre 1996 0 INTRODUCTION 1 DOMAINE D'APPLICATION 2 REFERENCES NORMATIVES 3 DEFINITIONS
PROBABILITES ET STATISTIQUE I&II
 PROBABILITES ET STATISTIQUE I&II TABLE DES MATIERES CHAPITRE I - COMBINATOIRE ELEMENTAIRE I.1. Rappel des notations de la théorie des ensemble I.1.a. Ensembles et sous-ensembles I.1.b. Diagrammes (dits
PROBABILITES ET STATISTIQUE I&II TABLE DES MATIERES CHAPITRE I - COMBINATOIRE ELEMENTAIRE I.1. Rappel des notations de la théorie des ensemble I.1.a. Ensembles et sous-ensembles I.1.b. Diagrammes (dits
L analyse d images regroupe plusieurs disciplines que l on classe en deux catégories :
 La vision nous permet de percevoir et d interpreter le monde qui nous entoure. La vision artificielle a pour but de reproduire certaines fonctionnalités de la vision humaine au travers de l analyse d images.
La vision nous permet de percevoir et d interpreter le monde qui nous entoure. La vision artificielle a pour but de reproduire certaines fonctionnalités de la vision humaine au travers de l analyse d images.
Document d orientation sur les allégations issues d essais de non-infériorité
 Document d orientation sur les allégations issues d essais de non-infériorité Février 2013 1 Liste de contrôle des essais de non-infériorité N o Liste de contrôle (les clients peuvent se servir de cette
Document d orientation sur les allégations issues d essais de non-infériorité Février 2013 1 Liste de contrôle des essais de non-infériorité N o Liste de contrôle (les clients peuvent se servir de cette
Rapport de certification
 Rapport de certification Memory Arrays avec Memory Gateways Version 5.5.2 Préparé par : Le Centre de la sécurité des télécommunications à titre d organisme de certification dans le cadre du Schéma canadien
Rapport de certification Memory Arrays avec Memory Gateways Version 5.5.2 Préparé par : Le Centre de la sécurité des télécommunications à titre d organisme de certification dans le cadre du Schéma canadien
PHYSIQUE-CHIMIE. Partie I - Spectrophotomètre à réseau
 PHYSIQUE-CHIMIE L absorption des radiations lumineuses par la matière dans le domaine s étendant du proche ultraviolet au très proche infrarouge a beaucoup d applications en analyse chimique quantitative
PHYSIQUE-CHIMIE L absorption des radiations lumineuses par la matière dans le domaine s étendant du proche ultraviolet au très proche infrarouge a beaucoup d applications en analyse chimique quantitative
NORME INTERNATIONALE
 NORME INTERNATIONALE ISO/CEl 1700 Première édition 1997-06-l 5 Technologies de l information - Interconnexion de systèmes ouverts (OSI) - Protocole de couche réseau ((Fast Byte» Information technology
NORME INTERNATIONALE ISO/CEl 1700 Première édition 1997-06-l 5 Technologies de l information - Interconnexion de systèmes ouverts (OSI) - Protocole de couche réseau ((Fast Byte» Information technology
Structures algébriques
 Structures algébriques 1. Lois de composition s Soit E un ensemble. Une loi de composition interne sur E est une application de E E dans E. Soient E et F deux ensembles. Une loi de composition externe
Structures algébriques 1. Lois de composition s Soit E un ensemble. Une loi de composition interne sur E est une application de E E dans E. Soient E et F deux ensembles. Une loi de composition externe
La segmentation à l aide de EG-SAS. A.Bouhia Analyste principal à la Banque Nationale du Canada. Chargé de cours à l UQAM
 La segmentation à l aide de EG-SAS A.Bouhia Analyste principal à la Banque Nationale du Canada. Chargé de cours à l UQAM Définition de la segmentation - Au lieu de considérer une population dans son ensemble,
La segmentation à l aide de EG-SAS A.Bouhia Analyste principal à la Banque Nationale du Canada. Chargé de cours à l UQAM Définition de la segmentation - Au lieu de considérer une population dans son ensemble,
Perrothon Sandrine UV Visible. Spectrophotométrie d'absorption moléculaire Étude et dosage de la vitamine B 6
 Spectrophotométrie d'absorption moléculaire Étude et dosage de la vitamine B 6 1 1.But et théorie: Le but de cette expérience est de comprendre l'intérêt de la spectrophotométrie d'absorption moléculaire
Spectrophotométrie d'absorption moléculaire Étude et dosage de la vitamine B 6 1 1.But et théorie: Le but de cette expérience est de comprendre l'intérêt de la spectrophotométrie d'absorption moléculaire
Ebauche Rapport finale
 Ebauche Rapport finale Sommaire : 1 - Introduction au C.D.N. 2 - Définition de la problématique 3 - Etat de l'art : Présentatio de 3 Topologies streaming p2p 1) INTRODUCTION au C.D.N. La croissance rapide
Ebauche Rapport finale Sommaire : 1 - Introduction au C.D.N. 2 - Définition de la problématique 3 - Etat de l'art : Présentatio de 3 Topologies streaming p2p 1) INTRODUCTION au C.D.N. La croissance rapide
Data Mining. Vincent Augusto 2012-2013. École Nationale Supérieure des Mines de Saint-Étienne. Data Mining. V. Augusto.
 des des Data Mining Vincent Augusto École Nationale Supérieure des Mines de Saint-Étienne 2012-2013 1/65 des des 1 2 des des 3 4 Post-traitement 5 représentation : 6 2/65 des des Définition générale Le
des des Data Mining Vincent Augusto École Nationale Supérieure des Mines de Saint-Étienne 2012-2013 1/65 des des 1 2 des des 3 4 Post-traitement 5 représentation : 6 2/65 des des Définition générale Le
Conseil économique et social
 NATIONS UNIES E Conseil économique et social Distr. GÉNÉRALE ECE/CES/GE.20/2008/3 12 février 2008 FRANÇAIS Original: ANGLAIS COMMISSION ÉCONOMIQUE POUR L EUROPE CONFÉRENCE DES STATISTICIENS EUROPÉENS Réunion
NATIONS UNIES E Conseil économique et social Distr. GÉNÉRALE ECE/CES/GE.20/2008/3 12 février 2008 FRANÇAIS Original: ANGLAIS COMMISSION ÉCONOMIQUE POUR L EUROPE CONFÉRENCE DES STATISTICIENS EUROPÉENS Réunion
La fonction exponentielle
 DERNIÈRE IMPRESSION LE 2 novembre 204 à :07 La fonction exponentielle Table des matières La fonction exponentielle 2. Définition et théorèmes.......................... 2.2 Approche graphique de la fonction
DERNIÈRE IMPRESSION LE 2 novembre 204 à :07 La fonction exponentielle Table des matières La fonction exponentielle 2. Définition et théorèmes.......................... 2.2 Approche graphique de la fonction
L utilisation d un réseau de neurones pour optimiser la gestion d un firewall
 L utilisation d un réseau de neurones pour optimiser la gestion d un firewall Réza Assadi et Karim Khattar École Polytechnique de Montréal Le 1 mai 2002 Résumé Les réseaux de neurones sont utilisés dans
L utilisation d un réseau de neurones pour optimiser la gestion d un firewall Réza Assadi et Karim Khattar École Polytechnique de Montréal Le 1 mai 2002 Résumé Les réseaux de neurones sont utilisés dans
- MANIP 2 - APPLICATION À LA MESURE DE LA VITESSE DE LA LUMIÈRE
 - MANIP 2 - - COÏNCIDENCES ET MESURES DE TEMPS - APPLICATION À LA MESURE DE LA VITESSE DE LA LUMIÈRE L objectif de cette manipulation est d effectuer une mesure de la vitesse de la lumière sur une «base
- MANIP 2 - - COÏNCIDENCES ET MESURES DE TEMPS - APPLICATION À LA MESURE DE LA VITESSE DE LA LUMIÈRE L objectif de cette manipulation est d effectuer une mesure de la vitesse de la lumière sur une «base
UE6 - Cycle de vie du médicament : Conception rationnelle
 UE6 - Cycle de vie du médicament : Conception rationnelle Dr. Raphaël Terreux Faculté de Pharmacie (ISPB) Département pédagogique des Sciences Physico-Chimiques et Pharmacie Galénique 8 avenue Rockefeller,
UE6 - Cycle de vie du médicament : Conception rationnelle Dr. Raphaël Terreux Faculté de Pharmacie (ISPB) Département pédagogique des Sciences Physico-Chimiques et Pharmacie Galénique 8 avenue Rockefeller,
Modélisation prédictive et incertitudes. P. Pernot. Laboratoire de Chimie Physique, CNRS/U-PSUD, Orsay [email protected]
 Modélisation prédictive et incertitudes P. Pernot Laboratoire de Chimie Physique, CNRS/U-PSUD, Orsay [email protected] Le concept de Mesure Virtuelle mesure virtuelle résultat d un modèle visant
Modélisation prédictive et incertitudes P. Pernot Laboratoire de Chimie Physique, CNRS/U-PSUD, Orsay [email protected] Le concept de Mesure Virtuelle mesure virtuelle résultat d un modèle visant
Formats 3D Critères d utilisation dans les échanges Frédéric CHAMBOLLE PSA Peugeot Citroën Direction des Systèmes d Information
 Formats 3D Critères d utilisation dans les échanges Frédéric CHAMBOLLE PSA Peugeot Citroën Direction des Systèmes d Information Atelier Ingénierie GALIA 30 novembre 2010 Introduction Les travaux de ce
Formats 3D Critères d utilisation dans les échanges Frédéric CHAMBOLLE PSA Peugeot Citroën Direction des Systèmes d Information Atelier Ingénierie GALIA 30 novembre 2010 Introduction Les travaux de ce
Créer le schéma relationnel d une base de données ACCESS
 Utilisation du SGBD ACCESS Polycopié réalisé par Chihab Hanachi et Jean-Marc Thévenin Créer le schéma relationnel d une base de données ACCESS GENERALITES SUR ACCESS... 1 A PROPOS DE L UTILISATION D ACCESS...
Utilisation du SGBD ACCESS Polycopié réalisé par Chihab Hanachi et Jean-Marc Thévenin Créer le schéma relationnel d une base de données ACCESS GENERALITES SUR ACCESS... 1 A PROPOS DE L UTILISATION D ACCESS...
ESSEC. Cours «Management bancaire» Séance 3 Le risque de crédit Le scoring
 ESSEC Cours «Management bancaire» Séance 3 Le risque de crédit Le scoring Les méthodes d évaluation du risque de crédit pour les PME et les ménages Caractéristiques Comme les montants des crédits et des
ESSEC Cours «Management bancaire» Séance 3 Le risque de crédit Le scoring Les méthodes d évaluation du risque de crédit pour les PME et les ménages Caractéristiques Comme les montants des crédits et des
Université de Lausanne
 Université de Lausanne Records management et archivage électronique : cadre normatif Page 2 Ce qui se conçoit bien s énonce clairement Nicolas Boileau Page 3 Table des matières Qu est- ce que le «records
Université de Lausanne Records management et archivage électronique : cadre normatif Page 2 Ce qui se conçoit bien s énonce clairement Nicolas Boileau Page 3 Table des matières Qu est- ce que le «records
1.2 Coordinence. Notion de liaison de coordinence : Cas de NH 3. et NH 4+ , 3 liaisons covalentes + 1 liaison de coordinence.
 Règle de l octet : tendance qu on les atomes à s entourer de 8 électrons dans l édifice moléculaire. Ce n est pas une règle générale. Composés respectant la règle de l octet Composés ne respectant pas
Règle de l octet : tendance qu on les atomes à s entourer de 8 électrons dans l édifice moléculaire. Ce n est pas une règle générale. Composés respectant la règle de l octet Composés ne respectant pas
Projet Active Object
 Projet Active Object TAO Livrable de conception et validation Romain GAIDIER Enseignant : M. Noël PLOUZEAU, ISTIC / IRISA Pierre-François LEFRANC Master 2 Informatique parcours MIAGE Méthodes Informatiques
Projet Active Object TAO Livrable de conception et validation Romain GAIDIER Enseignant : M. Noël PLOUZEAU, ISTIC / IRISA Pierre-François LEFRANC Master 2 Informatique parcours MIAGE Méthodes Informatiques
(Third-Man Attack) PASCAL BONHEUR PASCAL [email protected] 4/07/2001. Introduction. 1 Domain Name Server. 2 Commandes DNS. 3 Hacking des serveurs DNS
 PASCAL BONHEUR PASCAL BONHEUR@YAHOO.FR 4/07/2001. Introduction. 1 Domain Name Server. 2 Commandes DNS. 3 Hacking des serveurs DNS") Détournement de serveur DNS (Third-Man Attack) PASCAL BONHEUR PASCAL [email protected] 4/07/2001 Introduction Ce document traite de la possibilité d exploiter le serveur DNS pour pirater certains sites
Détournement de serveur DNS (Third-Man Attack) PASCAL BONHEUR PASCAL [email protected] 4/07/2001 Introduction Ce document traite de la possibilité d exploiter le serveur DNS pour pirater certains sites
Master IAD Module PS. Reconnaissance de la parole (suite) Alignement temporel et Programmation dynamique. Gaël RICHARD Février 2008
 Alignement temporel et Programmation dynamique. Gaël RICHARD Février 2008") Master IAD Module PS Reconnaissance de la parole (suite) Alignement temporel et Programmation dynamique Gaël RICHARD Février 2008 1 Reconnaissance de la parole Introduction Approches pour la reconnaissance
Master IAD Module PS Reconnaissance de la parole (suite) Alignement temporel et Programmation dynamique Gaël RICHARD Février 2008 1 Reconnaissance de la parole Introduction Approches pour la reconnaissance
Introduction MOSS 2007
 Introduction MOSS 2007 Z 2 Chapitre 01 Introduction à MOSS 2007 v. 1.0 Sommaire 1 SharePoint : Découverte... 3 1.1 Introduction... 3 1.2 Ce que vous gagnez à utiliser SharePoint... 3 1.3 Dans quel cas
Introduction MOSS 2007 Z 2 Chapitre 01 Introduction à MOSS 2007 v. 1.0 Sommaire 1 SharePoint : Découverte... 3 1.1 Introduction... 3 1.2 Ce que vous gagnez à utiliser SharePoint... 3 1.3 Dans quel cas
6. Hachage. Accès aux données d'une table avec un temps constant Utilisation d'une fonction pour le calcul d'adresses
 6. Hachage Accès aux données d'une table avec un temps constant Utilisation d'une fonction pour le calcul d'adresses PLAN Définition Fonctions de Hachage Méthodes de résolution de collisions Estimation
6. Hachage Accès aux données d'une table avec un temps constant Utilisation d'une fonction pour le calcul d'adresses PLAN Définition Fonctions de Hachage Méthodes de résolution de collisions Estimation
Cours Fonctions de deux variables
 Cours Fonctions de deux variables par Pierre Veuillez 1 Support théorique 1.1 Représentation Plan et espace : Grâce à un repère cartésien ( ) O, i, j du plan, les couples (x, y) de R 2 peuvent être représenté
Cours Fonctions de deux variables par Pierre Veuillez 1 Support théorique 1.1 Représentation Plan et espace : Grâce à un repère cartésien ( ) O, i, j du plan, les couples (x, y) de R 2 peuvent être représenté
Disparités entre les cantons dans tous les domaines examinés
 Office fédéral de la statistique Bundesamt für Statistik Ufficio federale di statistica Uffizi federal da statistica Swiss Federal Statistical Office EMBARGO: 02.05.2005, 11:00 COMMUNIQUÉ DE PRESSE MEDIENMITTEILUNG
Office fédéral de la statistique Bundesamt für Statistik Ufficio federale di statistica Uffizi federal da statistica Swiss Federal Statistical Office EMBARGO: 02.05.2005, 11:00 COMMUNIQUÉ DE PRESSE MEDIENMITTEILUNG
JOURNÉE THÉMATIQUE SUR LES RISQUES
 Survol de Risk IT UN NOUVEAU RÉFÉRENTIEL DE GESTION DES RISQUES TI GP - Québec 2010 JOURNÉE THÉMATIQUE SUR LES RISQUES 3 mars 2010 - Version 4.0 Mario Lapointe ing. MBA CISA CGEIT [email protected]
Survol de Risk IT UN NOUVEAU RÉFÉRENTIEL DE GESTION DES RISQUES TI GP - Québec 2010 JOURNÉE THÉMATIQUE SUR LES RISQUES 3 mars 2010 - Version 4.0 Mario Lapointe ing. MBA CISA CGEIT [email protected]
 Utilisation d informations visuelles dynamiques en asservissement visuel Armel Crétual IRISA, projet TEMIS puis VISTA L asservissement visuel géométrique Principe : Réalisation d une tâche robotique par
Utilisation d informations visuelles dynamiques en asservissement visuel Armel Crétual IRISA, projet TEMIS puis VISTA L asservissement visuel géométrique Principe : Réalisation d une tâche robotique par
Modélisation des données
 Modélisation des données Le modèle Entité/Association Le MCD ou modèle Entité/Association est un modèle chargé de représenter sous forme graphique les informations manipulées par le système (l entreprise)
Modélisation des données Le modèle Entité/Association Le MCD ou modèle Entité/Association est un modèle chargé de représenter sous forme graphique les informations manipulées par le système (l entreprise)
DETERMINATION DE L INCERTITUDE DE MESURE POUR LES ANALYSES CHIMIQUES QUANTITATIVES
 Agence fédérale pour la Sécurité de la Chaîne alimentaire Administration des Laboratoires Procédure DETERMINATION DE L INCERTITUDE DE MESURE POUR LES ANALYSES CHIMIQUES QUANTITATIVES Date de mise en application
Agence fédérale pour la Sécurité de la Chaîne alimentaire Administration des Laboratoires Procédure DETERMINATION DE L INCERTITUDE DE MESURE POUR LES ANALYSES CHIMIQUES QUANTITATIVES Date de mise en application
Projet de Traitement du Signal Segmentation d images SAR
 Projet de Traitement du Signal Segmentation d images SAR Introduction En analyse d images, la segmentation est une étape essentielle, préliminaire à des traitements de haut niveau tels que la classification,
Projet de Traitement du Signal Segmentation d images SAR Introduction En analyse d images, la segmentation est une étape essentielle, préliminaire à des traitements de haut niveau tels que la classification,
FORMATION CONTINUE SUR L UTILISATION D EXCEL DANS L ENSEIGNEMENT Expérience de l E.N.S de Tétouan (Maroc)
") 87 FORMATION CONTINUE SUR L UTILISATION D EXCEL DANS L ENSEIGNEMENT Expérience de l E.N.S de Tétouan (Maroc) Dans le cadre de la réforme pédagogique et de l intérêt que porte le Ministère de l Éducation
87 FORMATION CONTINUE SUR L UTILISATION D EXCEL DANS L ENSEIGNEMENT Expérience de l E.N.S de Tétouan (Maroc) Dans le cadre de la réforme pédagogique et de l intérêt que porte le Ministère de l Éducation
Nombres, mesures et incertitudes en sciences physiques et chimiques. Groupe des Sciences physiques et chimiques de l IGEN
 Nombres, mesures et incertitudes en sciences physiques et chimiques. Groupe des Sciences physiques et chimiques de l IGEN Table des matières. Introduction....3 Mesures et incertitudes en sciences physiques
Nombres, mesures et incertitudes en sciences physiques et chimiques. Groupe des Sciences physiques et chimiques de l IGEN Table des matières. Introduction....3 Mesures et incertitudes en sciences physiques
Extraction d informations stratégiques par Analyse en Composantes Principales
 Extraction d informations stratégiques par Analyse en Composantes Principales Bernard DOUSSET IRIT/ SIG, Université Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 04 [email protected] 1 Introduction
Extraction d informations stratégiques par Analyse en Composantes Principales Bernard DOUSSET IRIT/ SIG, Université Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 04 [email protected] 1 Introduction
Modèle Cobit www.ofppt.info
 ROYAUME DU MAROC Office de la Formation Professionnelle et de la Promotion du Travail Modèle Cobit DIRECTION RECHERCHE ET INGENIERIE DE FORMATION SECTEUR NTIC Sommaire 1. Introduction... 2 2. Chapitre
ROYAUME DU MAROC Office de la Formation Professionnelle et de la Promotion du Travail Modèle Cobit DIRECTION RECHERCHE ET INGENIERIE DE FORMATION SECTEUR NTIC Sommaire 1. Introduction... 2 2. Chapitre
EXERCICES : MECANISMES DE L IMMUNITE : pages 406 407 408 409 410
 EXERCICES : MECANISMES DE L IMMUNITE : pages 406 407 408 409 410 EXERCICE 1 PAGE 406 : EXPERIENCES A INTERPRETER Question : rôles respectifs du thymus et de la moelle osseuse dans la production des lymphocytes.
EXERCICES : MECANISMES DE L IMMUNITE : pages 406 407 408 409 410 EXERCICE 1 PAGE 406 : EXPERIENCES A INTERPRETER Question : rôles respectifs du thymus et de la moelle osseuse dans la production des lymphocytes.
Pourquoi l apprentissage?
 Pourquoi l apprentissage? Les SE sont basés sur la possibilité d extraire la connaissance d un expert sous forme de règles. Dépend fortement de la capacité à extraire et formaliser ces connaissances. Apprentissage
Pourquoi l apprentissage? Les SE sont basés sur la possibilité d extraire la connaissance d un expert sous forme de règles. Dépend fortement de la capacité à extraire et formaliser ces connaissances. Apprentissage
COMMUNICATEUR BLISS COMMANDE PAR UN SENSEUR DE POSITION DE L'OEIL
 COMMUNICATEUR BLISS COMMANDE PAR UN SENSEUR DE POSITION DE L'OEIL J. TICHON(1) (2), J.-M. TOULOTTE(1), G. TREHOU (1), H. DE ROP (2) 1. INTRODUCTION Notre objectif est de réaliser des systèmes de communication
COMMUNICATEUR BLISS COMMANDE PAR UN SENSEUR DE POSITION DE L'OEIL J. TICHON(1) (2), J.-M. TOULOTTE(1), G. TREHOU (1), H. DE ROP (2) 1. INTRODUCTION Notre objectif est de réaliser des systèmes de communication
Qualité du service et VoiP:
 Séminaire régional sur les coûts et tarifs pour les pays membres du Groupe AF Bamako (Mali), 7-9 avril 2003 1 Qualité du service et VoiP: Aperçu général et problèmes duvoip Mark Scanlan Aperçu général
Séminaire régional sur les coûts et tarifs pour les pays membres du Groupe AF Bamako (Mali), 7-9 avril 2003 1 Qualité du service et VoiP: Aperçu général et problèmes duvoip Mark Scanlan Aperçu général
Analyse,, Conception des Systèmes Informatiques
 Analyse,, Conception des Systèmes Informatiques Méthode Analyse Conception Introduction à UML Génie logiciel Définition «Ensemble de méthodes, techniques et outils pour la production et la maintenance
Analyse,, Conception des Systèmes Informatiques Méthode Analyse Conception Introduction à UML Génie logiciel Définition «Ensemble de méthodes, techniques et outils pour la production et la maintenance
Indications pour une progression au CM1 et au CM2
 Indications pour une progression au CM1 et au CM2 Objectif 1 Construire et utiliser de nouveaux nombres, plus précis que les entiers naturels pour mesurer les grandeurs continues. Introduction : Découvrir
Indications pour une progression au CM1 et au CM2 Objectif 1 Construire et utiliser de nouveaux nombres, plus précis que les entiers naturels pour mesurer les grandeurs continues. Introduction : Découvrir
Le Data Mining au service du Scoring ou notation statistique des emprunteurs!
 France Le Data Mining au service du Scoring ou notation statistique des emprunteurs! Comme le rappelle la CNIL dans sa délibération n 88-083 du 5 Juillet 1988 portant adoption d une recommandation relative
France Le Data Mining au service du Scoring ou notation statistique des emprunteurs! Comme le rappelle la CNIL dans sa délibération n 88-083 du 5 Juillet 1988 portant adoption d une recommandation relative
Faculté des sciences de gestion et sciences économiques BASE DE DONNEES
 BASE DE DONNEES La plupart des entreprises possèdent des bases de données informatiques contenant des informations essentielles à leur fonctionnement. Ces informations concernent ses clients, ses produits,
BASE DE DONNEES La plupart des entreprises possèdent des bases de données informatiques contenant des informations essentielles à leur fonctionnement. Ces informations concernent ses clients, ses produits,
NORME 4 SYSTÈME DE CLASSIFICATION DES PERMIS DE CONDUIRE
 NORME 4 YTÈME DE CLAIFICATION DE PERMI DE CONDUIRE Bien que cette Norme apparaisse dans le Code canadien de sécurité pour les transporteurs routiers, il est important de noter qu'elle s'applique à tous
NORME 4 YTÈME DE CLAIFICATION DE PERMI DE CONDUIRE Bien que cette Norme apparaisse dans le Code canadien de sécurité pour les transporteurs routiers, il est important de noter qu'elle s'applique à tous
ORACLE PRIMAVERA PORTFOLIO MANAGEMENT
 ORACLE PRIMAVERA PORTFOLIO MANAGEMENT FONCTIONNALITÉS GESTION DE PORTEFEUILLE Stratégie d approche permettant de sélectionner les investissements les plus rentables et de créer de la valeur Paramètres
ORACLE PRIMAVERA PORTFOLIO MANAGEMENT FONCTIONNALITÉS GESTION DE PORTEFEUILLE Stratégie d approche permettant de sélectionner les investissements les plus rentables et de créer de la valeur Paramètres