La Recherche clinique est essentielle
|
|
|
- Colette Fleury
- il y a 10 ans
- Total affichages :
Transcription
1 La Recherche clinique est essentielle Les essais clinique en Tunisie Point de vue du clinicien Professeur H. Haouala Tunis, le 09 février 2012 XVèmes Journées Pharmaceutiques Tunisiennes 1

2 Sommaire de la présentation 1. Généralités sur la recherche clinique : 2. Les différentes phases d études cliniques 3. La réglementation internationale 4. Les différentes étapes du suivi d une étude 5. La Recherche clinique en Tunisie 6. Rôle et mission de la CRO 2

3 1. Généralités sur la recherche clinique 3

4 Pourquoi tout cela a vu le jour Parce que même s il n existe pas de sciences sans test et donc sans preuves, on ne peut pas faire de tests sans les entourer de toute la rigueur scientifique, éthique et réglementaire. Car dans le passé, beaucoup ont fait n importe quoi Quelques exemples : 4

5 Exemples de l histoire «the Tuskegee Syphilis study»: étude sur des hommes noirs sans consentement éclairé Crimes de guerre (histoire du nazisme) «the Willowbrook study»: étude sur des enfants déficients mentaux délibérément infectés par le virus de l hépatite Distilbène : encore d actualité! Hormone de croissance 5

6 La Recherche Clinique, qu'est ce que c'est? C'est l'étape faisant suite à la recherche fondamentale et à l'expérimentation animale. Le délai moyen entre une découverte fondamentale et la commercialisation d un médicament est de 10 à 15 ans La recherche clinique est réalisée sur l'homme par des équipes qualifiées de médecins, de pharmaciens, de laborantins, d'infirmiers et d'autres personnels de santé. Elle se fait le plus souvent à l'hôpital, mais se fait également en ville Elle est prise en charge de plus en plus par des sociétés privées (CRO ou Contract Research Organization), qui sont des prestataires de l'industrie pharmaceutique. 6

7 Le coût de la recherche clinique Le programme de développement d un nouveau médicament coûte en moyenne au firmes pharmaceutiques entre 2 et 3 milliards de dollars Autant les phases précoces : fondamental, pré clinique, phase I et phase II peuvent concerner très peu de centres, autant les études pivotales de phase III nécessitent la mise en place de centaines de centres pour pouvoir recruter les milliers de patients requis. Il faut donc plusieurs dizaines de pays pour pouvoir finaliser le programme dans des temps corrects. 7

8 Définition d un essai clinique Un essai clinique est une étude médicale organisée pour tester les effets d'un nouveau médicament ou d'un médicament déjà existant, d'un traitement biologique, ou d'un dispositif médical qui pourrait traiter ou empêcher une maladie déjà identifiée. 8

9 Les essais cliniques Les études cliniques sont donc réalisées pour examiner, détecter, contrôler, traiter, ou prévenir toute maladie. L'objectif principal d'un essai clinique est de comparer 2 ou plusieurs groupes de patients, en utilisant 2 ou plusieurs traitements afin de déterminer l'efficacité d'un médicament ou d'un traitement biologique. 9

10 Les essais cliniques Les essais cliniques sont menés avec précaution et en respectant l éthique: - pour protéger les patients d'effets secondaires inutiles - pour permettre une collecte ainsi qu une analyse précise des données relatives à une maladie donnée. Les essais cliniques offrent l'espoir aux patients d'un traitement efficace contre leur maladie 10

11 2. Les différentes Phases d études 11

12 Les différentes Phases Pour répondre à ces différentes questions, les essais cliniques sont habituellement réalisés en 4 phases, appelées phases I, II, III, IV, permettant aux compagnies pharmaceutiques d'initier et de suivre le développement d'une nouvelle molécule ou d'un nouveau traitement. 12

13 Phase Préclinique La phase préclinique, qui se déroule in vitro et in vivo chez l'animal, comporte : - des études pharmacocinétiques (absorption, diffusion, élimination du médicament étudié), - des études de tolérance - des études pharmacodynamiques (recherche de dose). Ces études sont indispensables avant d'envisager l'administration du médicament chez l'être humain. 13

14 Phase I Ces études cliniques servent à évaluer l'innocuité et la pharmacocinétique (quantité et disponibilité dans l'organisme) des médicaments en développement. On étudie la tolérance en fonction de la dose et le métabolisme du médicament. Généralement, ce sont de petites études durant quelques jours à quelques semaines et impliquant un petit nombre de volontaires sains (personnes sans maladie diagnostiquée, désireuses de participer à une étude clinique). Les personnes sont souvent hospitalisées pendant l'étude pour être suivies rigoureusement. Cela est réalisé le plus souvent dans des unités spécialisés, appelées centre de phase I 14
. Les personnes sont souvent hospitalisées pendant l'étude pour être suivies rigoureusement.")
15 Phase II Les études cliniques de phase II, s'adressent généralement à des personnes malades (groupe homogène de quelques dizaines de malades ) et permettent de mettre en évidence l'efficacité du médicament. Durée : de quelques mois à 2 ans La toxicité avec la survenue éventuelle d'effets secondaires est également suivie scrupuleusement, afin d'évaluer le meilleur compromis efficacité/tolérance pour le patient. 15

16 Phase III Ces études sont généralement randomisées (attribution d'un numéro de traitement pour chaque patient donné) afin de contrôler avec précision les effets du médicament étudié. Ces études peuvent également comparer le médicament étudié aux traitements habituellement utilisés. 16

17 Phase III Les études cliniques dites de phase III regroupent un grand nombre de personnes malades (le groupe de malades étant moins homogène que dans les phases précédentes). Elles permettent de recueillir plus d'informations sur l'efficacité et la tolérance du médicament en développement, tout en le comparant aux médicaments habituellement utilisés, ou s'il n'en existe pas, à un placebo Ces études à grande échelle se déroulent la plupart du temps sur de longues périodes (plusieurs mois à plusieurs années). 17

18 Phase IV : Études cliniques post-amm Les études de phase IV se déroulent lorsque le médicament est commercialisé. Les médicaments utilisés au cours des études cliniques de phase IV ne peuvent être utilisés que dans l'indication prévue par l'amm. En dehors de l AMM, cela redevient une phase III. Elles regroupent un très grand nombre (plusieurs centaines à plusieurs milliers) de personnes malades dans un ou plusieurs pays. Elles permettent d'accumuler, d'une part une plus large connaissance sur les effets secondaires et l'efficacité liés au médicament, et d'autre part des informations pratiques (choix de la dose et de la formulation du traitement, traitement d'une autre maladie, ). 18
 de personnes malades dans un ou plusieurs pays.")
19 Phase IV : Enquêtes - Études non interventionnelles On ne parle plus d'étude clinique de médicament, mais d'étude observationnelle post-amm, avec ou sans prescription de médicament (enquête pharmaco économique, épidémiologique,...). Les enquêtes ont pour intérêt principal l'observation, la documentation des pratiques médicales et/ou l'évaluation dans la pratique courante du rapport efficacité/tolérance de médicament(s). Elles permettent de documenter l'efficacité, du risque et de l'usage des médicaments lors de la constitution des dossiers de renouvellement des AMM 19
.")
20 Différences entre phase IV et phase IV! Après l AMM, on peut réaliser, Soit: Une étude clinique de phase IV qui est menée exactement comme une phase II ou III. Soit: Dans une étude de phase IV, le produit utilisé est une Unité de Traitement mis à disposition gratuitement par le promoteur Une enquête ou une étude observationnelle, dans laquelle, on ne teste pas, on observe. Si le médecin a décidé de traiter un patient, avec un produit qu il a prescrit de son libre arbitre, on peut observer un certain nombre de données résumées dans un protocole.(ce type d étude est familièrement appelé, les phases V) 20

21 Une étude observationnelle, cela ne sert à rien? Cela peut en effet ne servir qu à pousser les médecins à prescrire Mais, bien faite, cela permet d apporter des informations fondamentales, notamment en terme: D épidémiologie, De détermination de spécificité loco-régionale, De détermination des différentes modalités de prise en charge, De détermination des différentes modalités de suivi, De formation médicale continue, De surveillance de la bonne utilisation des médicaments, et/ou des recommandations. L intérêt National est certain 21
22 3. La réglementation internationale 22
23 Les Dates-clés 1931 Food & Drug Administration (USA-FDA) 1947 Code de Nuremberg 1964 Déclaration d Helsinki 1978 Loi Informatique et Liberté (CNIL) 1983 Création du Comité National d Éthique 1987 Bonnes Pratiques Cliniques France 1988 Loi Huriet Sérusclat 1989 Japanese GCP law 1991 European Union -GCP guidelines 1997 ICH-GCP guidelines 1995 Directive EU sur la protection des données 2001 Directive européenne 2001/20/CE 23
24 La déclaration d Helsinki C est un ensemble de recommandations faites par l Association Médicale Mondiale (the World Medical Association) à l intention des médecins dans le domaine de la recherche biomédicale portant sur des sujets humains. Elle a été signée à Helsinki en juin 1964 et amendée en octobre 1975, octobre 1983 et septembre 1989, Octobre 1996, Octobre 2000 et lors de l'assemblée générale de l'amm à Washington en
25 Un texte éthique Assemblée Médicale Mondiale Déclaration d Helsinki : 1964 amendée en 2000 Un protocole Un comité d éthique indépendant Un médecin pour suivre les patients Un consentement éclairé La publication des résultats 25
26 La déclaration d Helsinki : Dernière version Octobre 2000 Prévenir et stopper l exploitation des populations pauvres par l industrie pharmaceutique et les instituts de recherche L accès au traitement du patient ayant participé à un essai clinique doit être garanti Les patients doivent toujours bénéficier du meilleur traitement 26
27 Loi Huriet Référence Française de 1989 à 2006 Sanctions pénales Dispositions générales du consentement Dispositions particulières Dispositions administratives 27
28 I.C.H-GCP EUROPE USA I.C.H JAPON Objectif : harmoniser les guidelines 28
29 ICH GCP International Conference on Harmonization of technical requirements for registration of pharmaceuticals for human use. Harmonized Tripartite Guidelines for Good Clinical Practice Membres et initiateurs : FDA EMEA MoH Japan 29
30 Pourquoi des BPC - ICH? Pour harmoniser les exigences techniques d enregistrement du médicament Pour garantir la protection des droits, de la sécurité et du bien-être des sujets dont les intérêts prévalent sur ceux de la science et de la société Pour garantir la crédibilité des résultats (documentés, accessibles, vérifiables) Pour répondre aux mêmes exigences techniques de qualité, d efficacité et de tolérance 30
31 ICH - BPC Les régions : L Union Européenne Japon USA Autres : Canada, Australie.. Mise en application : Toute étude démarrant après janvier
32 Sommaire ICH : 8 chapitres 1 : Glossaire 2 : 13 principes 3 : Comité d Éthique indépendant 4 : L investigateur 5 : Promoteur 6 : Protocole et amendements 7 : Brochure de l investigateur 8 : Documents essentiels 32
33 Les 13 principes ICH 1. Les études doivent être menées en accord avec les principes éthiques de la déclaration d Helsinki 2. S assurer avant le démarrage de l étude que les avantages sont supérieurs aux risques et inconvénients 3. Le droit, la sécurité et le bien être des patients sont les plus importantes considérations et doivent prévaloir tout au long de l étude 4. Les informations cliniques et non cliniques disponibles doivent être suffisantes pour proposer l étude 5. L étude doit être rigoureusement scientifique et décrite dans un protocole claire. 6. L étude doit être conduite en accord avec le protocole ayant était préalablement validé par un IRB (Institutional Review Board) et approuvé par un comité d éthique indépendant (Independant Ethical Committee) 33 33
34 Les 13 principes ICH 7. La prise en charge médicale et l ensemble des décisions médicales doivent être prises par un médecin qualifié. 8. Toute personne menant une étude doit être qualifiée par une formation, un training et une expérience, dans la conduite d une étude clinique. 9. Un libre consentement du patient doit être obtenu avant toute participation à l étude. 10. Toutes les données cliniques relatives à l étude doivent être enregistrées, manipulées et stockées d une manière permettant leur reporting, leur interprétation et leur vérification. 11. La confidentialité des données patients doit être totale 12. Les traitements doivent être fabriqués, manipulés et stockés en accord avec les principes de bonne fabrication 13. Un système qualité doit être implémenté 34
35 Comité d éthique selon ICH Responsabilités du CE : Doit assurer les droits, la sécurité et le bien être des patients dans l étude Doit exiger de recevoir le protocole, les amendements, l information et le consentement écrit, la brochure investigateur, CV des investigateurs S assurer de la qualification des investigateurs S assurer des aspects éthiques de la recherche S assurer que tout ce que va subir le patient est clairement mentionné dans le consentement et l information patient 35
36 Comité d éthique selon ICH Composition et règles de fonctionnement : Personnes expérimentées Au moins 5 membres Au moins un membre dont le premier centre d intérêt est sans rapport avec la recherche Au moins un membre n exerçant aucune activité dans l institution ou le site sur lequel est prévu la recherche Seuls les membres ne participants pas directement à la recherche doivent exprimer leur opinion Avis rendu si quorum atteint Aucun membre représentant le promoteur ou la CRO ne doit participer à la réunion sauf convocation pour demande d information 36
37 L investigateur selon ICH Formation et aptitudes : Doit être formé, habilité et expérimenté tant dans la réalisation d une étude clinique, que dans la pathologie étudiée Doit être formé aux GCP-ICH et à la réglementation en cours dans le pays Doit permettre le monitoring et les audits sur son centre et donc mettre à disposition les dossiers sources concernés Doit avoir une liste de personnes habilitées à qui il peut déléguer certaines parties de la recherche Doit être disposé à démontrer qu il a le potentiel de recrutement Doit disposer du temps nécessaire à la conduite d une étude Doit avoir un staff suffisant et disponible Doit s assurer que l ensemble des personnes participant à la recherche ont été formées 37
38 L investigateur selon ICH Doit être le seul responsable de l ensemble des décisions médicales Doit s assurer de la bonne prise en charge médicale Doit informer son correspondant MG si applicable Doit s assurer qu il existe un avis favorable du comité d éthique Doit remettre les versions mises à jour des documents au comité d éthique Doit se rendre disponible pour répondre aux éventuelles questions du comité d éthique Doit respecter scrupuleusement le protocole; Ne doit faire aucune déviation sans l accord du promoteur et si telle est le cas, il doit compléter une «deviation form» 38
39 L investigateur selon ICH L investigateur et les Unités de Traitements (UT) : S assurer que les traitements utilisés sont bien étiquetés «étude clinique» et à ce titre être totalement gratuits pour le patient et mis à disposition gratuitement par le Promoteur Responsable de la comptabilité des produits à l étude Le stockage des UT doit être en accord avec les conditions fixées par le promoteur Dois s assurer que les produits sont utilisés uniquement dans le cadre du protocole. Doit respecter les procédures de randomisation 39
40 L investigateur selon ICH Information et consentement patient : S assurer que les 2 éléments ont été approuvés par le comité d éthique S assurer que l information patient soit modifiée si le protocole a subit des modifications Ni l investigateur ni son staff ne doivent influencer un patient ne souhaitant pas participer. Explications, oui, autre chose : non Prendre le temps d expliquer au patient ou à son représentant légal l ensemble de l étude, objectifs, contraintes et intérêt Nécessité d avoir un consentement dans la langue natale du patient 40
41 L investigateur selon ICH Information et consentement patient : Expliquer au patient qu il a le droit de retirer en cours d étude son consentement sans que ce dernier ne soit dans l obligation d en donner les raisons et sans que cela ne change sa relation médecin-malade. S assurer que le patient a les facultés nécessaires pour comprendre les documents et suivre l étude. S assurer qu il a eu le temps de lire (il lui aura remis une copie au préalable) Obtenir son consentement éclairé, daté et signé et en remettre une copie au patient 41
42 L investigateur selon ICH Les données : Doit s assurer de l exactitude des informations qu il inscrit dans le Cahier d observation (CRF) ou cahier de receuil des données S assurer que les données reportées sur le CRF sont identiques à celles qu il a reporté sur son dossier source Toute modification sur le CRF doit d être datée et signée par l investigateur. Il doit rayer la notion fausse mais s assurer qu elle reste visible. 42
43 L investigateur selon ICH La tolérance : Tous les Evénements Indésirables Graves (EIG) doivent être rapportés immédiatement au promoteur Il doit compléter le formulaire adéquat Les Evénements Indésirables (EI) et les résultats biologiques anormaux doivent être rapportés sur le CRF sans délai spécifique Pour les décès et certains SAEs l investigateur sera amené à répondre aux demandes des autorités et/ou du sponsor Arrêt prématuré de l étude : Il devra en informer le patient et prendre les meilleures dispositions pour assurer la continuité du traitement Il devra s assurer que le comité d éthique soit informé 43
44 4. Les différentes étapes de suivi d une étude 44
45 Les étapes Sélection des investigateurs Visite de Mise en Place (précédée ou non d une réunion d investigateurs) Visite de suivi ou de monitoring Visite de Clôture 45
46 La sélection des investigateurs Objectif Trouver les médecins investigateurs pouvant réaliser l étude dans les meilleures conditions Médecins formés Médecins entourés d une équipe formée susceptible d aider l investigateur dans le suivi du patient dans le cadre de l étude Médecin ayant le potentiel de recrutement des patients concernés par l étude Médecin bénéficiant de l infrastructure rendue nécessaire par l étude 46
47 Déroulement de la mise en place Confirmer l'engagement de l'investigateur Vérifier la compréhension du protocole par les différents intervenants Détailler aux différents intervenants (investigateurs, infirmiers, pharmacien, ) les points essentiels du protocole, et du cahier d'observation Rappeler aux intervenants leurs obligations Contrôler la réception du matériel Récupérer les documents essentiels Détailler la procédure de déclaration des EI 47
48 LA MISE EN PLACE : Principe Sitôt la visite terminée, l'investigateur doit être en mesure de pouvoir inclure des patients. 48
49 Éléments expliqués lors de la MEP Le protocole : lecture détaillée du protocole Le cahier d'observation Le recueil obligatoire du consentement éclairé écrit des patients La comptabilité des unités thérapeutiques : le processus de dispensation et de récupération des unités thérapeutiques L'allocation des traitements (randomisation ou non) La gestion des enveloppes de randomisation 49
50 Éléments expliqués lors de la MEP Les traitements et les maladies concomitants et associés La procédure à suivre en cas de survenue d'événements indésirables Les Bonnes Pratiques Cliniques (Accès direct aux documents sources, Liste d'identification des patients, Visites des ARCs et correction des données, Levée d'anonymat, déclaration des EIG, Audit / Inspection, Archivage ) 50
51 Documentations MEP Le contrat signé d'expérimentation clinique Les conventions financières «Investigateur» Le protocole signé Le curriculum vitae daté et signé de(s) investigateur(s) et du ou des co investigateur(s) L'accusé de réception des unités thérapeutiques Valeurs normales du laboratoire Le certificat d'accréditation du laboratoire 51
52 MEP : matériel remis par l ARC Le dossier investigateur regroupant l'ensemble de la documentation du centre, entre autres : La brochure investigateur Avis favorable du CCPPRB Avis favorable de la DPM en Tunisie L'attestation d'assurance Les formulaires d'information et de consentement Les textes (Loi Huriet, déclaration d Helsinki, ICH) Les cahiers d'observation Les unités thérapeutiques Les bulletins d'analyse des unités thérapeutiques Les enveloppes de randomisation / décodage 52
53 Visite de suivi ou de monitoring : L ARC vérifie Le bon déroulement de l'inclusion des patients Le respect des critères de sélection Le respect de la chronologie d'allocation des traitements en cas d'études comparatives Le respect de la chronologie des évaluations, des examens,... Le remplissage de l'ensemble des variables du cahier d'observation La cohérence des données reportées dans le cahier d'observation 53
54 Visite de suivi ou de monitoring : L ARC vérifie La conformité entre les données reportées dans le cahier d'observation et celles figurant dans le document source (Vérifier l'authenticité et la fiabilité des données recueillies) Le remplissage de la feuille de comptabilité des unités thérapeutiques La transmission en temps utile des événements indésirables au promoteur 54
55 Visite de suivi ou de monitoring : principe La première visite de suivi dans un centre doit avoir lieu le plus tôt possible après la première inclusion 55
56 Points abordés lors des visites de suivi L'avancement de l'étude Les consentements éclairés Les cahiers d'observation Le remplissage - report des données : Les données reportées doivent être identiques à celle des documents sources Les événements indésirables graves survenus depuis la visite précédente Les unités thérapeutiques (stockage et comptabilité) 56
57 Points abordés lors des visites de suivi Avancement de l'étude Le nombre de patients : Prévus Sélectionnés Inclus Inclus à tort Ayant achevés l'étude Prématurément arrêtés Perdus de vue Les difficultés éventuellement rencontrées 57
58 Vérification du CRF Absence de pages manquantes, numéro du patient, du centre, Signature, lisibilité Absence de données manquantes, nombre de chiffres après la virgule La cohérence des données croisées entre elles Seul le ou les investigateur(s) ou les co-investigateurs sont habilités à inscrire et signer les corrections Respect du protocole 58
59 Vérification du CRF La méthodologie des corrections : l'investigateur barre la donnée erronée tout en la laissant encore lisible. La correction est datée et signée. Des demandes de corrections peuvent aussi être fournies par le service du data management par l'intermédiaire de formulaires de correction ou «queries». Unité des variables numériques, absence d'anomalie dans l'évolution des critères, valeurs biologiques dans la limite acceptable, cohérence entre les réponses. 59
60 Vérification du CRF Respect des critères de sélection, respect des délais entre les évaluations, les prises de traitement, précision des mesures du critère principal, respect du schéma thérapeutique, suivi correct des événements indésirables. Vérification de la survenue d'événements indésirables graves survenus depuis la dernière visite, dans l'affirmative vérifier si ceux-ci ont été déclarés et documentés. 60
61 Vérification Consentements éclairés en 3 exemplaires : Patient Investigateur Enveloppe scellée à destination du promoteur Unités thérapeutiques et Enveloppes de randomisation Vérification de l'allocation des traitements Vérification du stock, de la feuille de comptabilité Vérification des enveloppes de randomisation ou du respect du système de codage 61
62 La visite de clôture Elle met un terme au travail d'investigation et de monitoring. L ARC s assure : que tous les cahiers d'observation ainsi que les bordereaux de correction ont été complétés de la réexpédition des traitements du respect des obligations d'archivage 62
63 Fin normale de l essai Lorsque les données recueillies dans les CRF sont cohérentes, complètes ou définitivement irrécupérables (décision biométricien et/ou CP) 63
64 Clôture prématurée Les raisons doivent avoir été prévues dans le protocole. C est une décision du promoteur, des autorités ou de l'investigateur. Les raisons d'une clôture prématurée peuvent être: absence de recrutement retard dans le recrutement non respect du protocole survenue d'eig entraînant l'arrêt de l'essai inefficacité évidente du médicament succès majeur du médicament (analyse intermédiaire) 64
65 Points à aborder lors de la clôture Bilan de l'étude: Le nombre de patients prévus, sélectionnés, inclus, inclus à tort, ayant achevés l'étude, prématurément arrêtés et perdus de vue Cahiers d'observation: Correction des originaux, possibilité d'avoir des demandes de correction Comptabilités des cahiers complétés, vierges Nouvelle vérification des consentements (absence de retrait) 65
66 Points à aborder lors de la clôture Unités thérapeutiques et enveloppes de randomisation : Décompte de toutes les unités thérapeutiques, Vérifier que la feuille de comptabilité des unités thérapeutiques est à jour et signée par l'investigateur Décompte des conditionnements non utilisés à faire expédier au promoteur, Décompte des enveloppes de randomisation Matériel 66
67 5. La recherche clinique en Tunisie Intérêts, forces et faiblesses.. 67
68 Intérêt Pour le pays et les autorités : Faire rentrer la Tunisie au firmament des pays participants au développement clinique internationale Permettre aux autorités en charge de l enregistrement des produits d avoir une vision précoce et très développé du dossier d enregistrement Permettre aux autorités en charge de l enregistrement de pouvoir bénéficier de conseils d experts nationaux maitrisant le développement de la molécule Augmenter le nombre d experts Tunisiens participant à la signature d articles indexés Voir un certain nombre de prise en charge thérapeutiques patients assurer par les sponsors d études cliniques Essayer de négocier avec les sponsors des programmes de prise en charge thérapeutiques à objet compassionnel 68
69 Intérêts Pour le médecin : Acteur, plus qu observateur Connaissance en amont plutôt qu en aval Ecole de la rigueur, car par définition un protocole est une somme d exigences Suivi obligatoire plus intense que le suivi habituel Meilleure prise en compte de l importance du dossier patient Meilleure prise en compte des nécessités de suivi 69
70 Intérêts Pour le patient : Meilleure connaissance de sa pathologie Meilleure connaissance de son traitement Relation plus étroite avec son médecin Prise en charge générale de l ensemble des frais liés à son traitement Bénéficie d examens et de suivi le plus souvent plus approfondis 70
71 L audit L'audit est un processus systématique, indépendant et documenté permettant de recueillir des informations objectives pour déterminer dans quelle mesure les éléments du système cible satisfont aux exigences des référentiels du domaine concerné. Dans le cadre de la recherche clinique, l audit un processus de contrôle qualité qui a pour objectif de vérifier le respect des procédures de l étude. Cet audit peut être déclenché : Avant l étude : pour voir si le centre est apte à conduire l étude prévue Pendant l étude A la fin de l étude L audit peut être déclenché par : Les autorités réglementaires : DPM en Tunsie/AFSSAPS en France, EMEA en Europe, FDA aux US Le laboratoire sponsor 71
72 L audit L audit est conduit par un auditeur qualité indépendant Il vérifiera sur site l ensemble des élements liés à l étude et ce sur les aspects suivants : Vérification du dossier réglementaire : avis des CE, des autorités de santé, des soumissions des amendements, des contrats Staff log et CV des investigateurs et co investigateurs Vérification des signatures des informations et consentements patients Respect des procédures de l étude Vérification de la réelle existence des patients Respect des conditions d étiquetage et de délivrance des unités de traitement Comptage des unités de traitements Vérification des dossiers patients Vérification des conditions de confidentialité.. 72
73 Tunisie et recherche clinique La Tunisie jouit déjà d une expérience certaine dans le domaine de la recherche clinique Même si le nombre d études réalisés reste très faible, (en moyenne une dizaine par an), la Tunisie malgré sa taille est le 3 eme pays Africain en nombre d études après l Afrique du sud (loin devant) et l Egypte 73
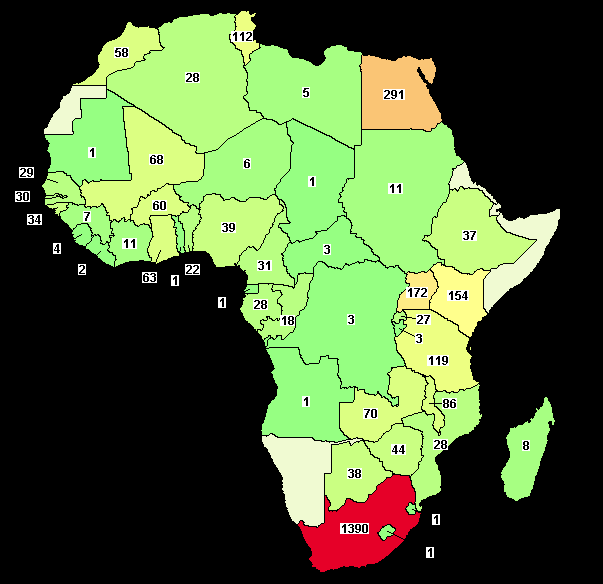
74 74
75 75
76 Les forces Une infrastructure hospitalière des plus développée Un potentiel de recrutement de patients optimale Des plateaux techniques permettant la conduite de tout type d études Des médecins investigateurs qui pour certains ont déjà un niveau d expérience et de formation des plus corrects Une réglementation collant dans sa grande majorité au prérequis des BPC-ICH Des comités d éthiques présents dans la majorité des grands établissements 76
77 Les faiblesses Un process de déclaration des événements de pharmacovigilance à optimiser Un défaut de récupération de rapports de fin d études et des rapports de pharmacovigilance tant pour la DPM que pour les comités d éthique. L absence d information des pharmacies hospitalières L absence de réglementation pour les études épidémiologiques observationnelles 77
Estelle Marcault. 20/01/2012 URC Paris Nord 1
 Estelle Marcault 20/01/2012 URC Paris Nord 1 Définition du Monitoring Surveillance de l avancement d un essai clinique Garantie que la conduite de l essai, les enregistrements et les rapports sont réalisés
Estelle Marcault 20/01/2012 URC Paris Nord 1 Définition du Monitoring Surveillance de l avancement d un essai clinique Garantie que la conduite de l essai, les enregistrements et les rapports sont réalisés
OUVERTURE ET MISE EN PLACE
 OUVERTURE ET MISE EN PLACE Estelle Marcault 20/01/2012 URC PARIS NORD 1 Ouverture et mise en place Trois types de visites/ réunions peuvent avoir lieu : Visite de sélection Réunion investigateur Visite
OUVERTURE ET MISE EN PLACE Estelle Marcault 20/01/2012 URC PARIS NORD 1 Ouverture et mise en place Trois types de visites/ réunions peuvent avoir lieu : Visite de sélection Réunion investigateur Visite
MONITORING / SUIVI DES PATIENTS
 Formation Recherche Clinique OncoBasseNormandie 02/12/2013 MONITORING / SUIVI DES PATIENTS Jean-Michel GRELLARD ARC Coordinateur - Centre François Baclesse Quelques définitions Cahier d observation ou
Formation Recherche Clinique OncoBasseNormandie 02/12/2013 MONITORING / SUIVI DES PATIENTS Jean-Michel GRELLARD ARC Coordinateur - Centre François Baclesse Quelques définitions Cahier d observation ou
Audit et Inspection Les contraintes extérieures B.Malivoir
 Audit et Inspection Les contraintes extérieures B.Malivoir Chef de projet Hémato-Onco CHRU Tours Vice Présidente CPP Région Ouest1 Le contexte juridique Directive 2001/20/CE du Parlement européen et du
Audit et Inspection Les contraintes extérieures B.Malivoir Chef de projet Hémato-Onco CHRU Tours Vice Présidente CPP Région Ouest1 Le contexte juridique Directive 2001/20/CE du Parlement européen et du
Unité de Recherche Clinique St Louis - Lariboisière Fernand Widal Le 03 Février 2012
 Visite de pré-sélection Visite de Mise en place Murielle COURREGES-VIAUD, ARC Laurence GUERY, ARC responsable Assurance Qualité Véronique JOUIS, Coordinatrice des ARCs Responsable Logistique Unité de Recherche
Visite de pré-sélection Visite de Mise en place Murielle COURREGES-VIAUD, ARC Laurence GUERY, ARC responsable Assurance Qualité Véronique JOUIS, Coordinatrice des ARCs Responsable Logistique Unité de Recherche
Introduction au métier d ARC. en recherche clinique
 Introduction au métier d ARC en recherche clinique Déroulement d un projet de recherche clinique Idée Faisabilité Avant Pendant Après Protocole accepté Démarches réglementaires Déroulement de l étude Analyse
Introduction au métier d ARC en recherche clinique Déroulement d un projet de recherche clinique Idée Faisabilité Avant Pendant Après Protocole accepté Démarches réglementaires Déroulement de l étude Analyse
Rôle de l Assurance Qualité dans la recherche clinique
 Rôle de l Assurance Qualité dans la recherche clinique Pôle Qualité / Gestion des Risques 05 Janvier 2012 Plan La qualité d une recherche c est quoi? Bonnes Pratiques Cliniques (BPC) Responsabilités des
Rôle de l Assurance Qualité dans la recherche clinique Pôle Qualité / Gestion des Risques 05 Janvier 2012 Plan La qualité d une recherche c est quoi? Bonnes Pratiques Cliniques (BPC) Responsabilités des
LES ETUDES CLINIQUES EN 20 QUESTIONS
 LES ETUDES CLINIQUES EN 20 QUESTIONS La mise au point d un nouveau médicament est longue. Sur environ 10 000 médicaments potentiels subissant tous les tests nécessaires, un seul sera disponible au final
LES ETUDES CLINIQUES EN 20 QUESTIONS La mise au point d un nouveau médicament est longue. Sur environ 10 000 médicaments potentiels subissant tous les tests nécessaires, un seul sera disponible au final
Estelle Marcault 06/02/2015 URC PARIS NORD 1
 Estelle Marcault 06/02/2015 URC PARIS NORD 1 Définition du Monitoring Garantie que la conduite de l essai clinique, les enregistrements et les rapports sont réalisés conformément : Au protocole Aux Procédures
Estelle Marcault 06/02/2015 URC PARIS NORD 1 Définition du Monitoring Garantie que la conduite de l essai clinique, les enregistrements et les rapports sont réalisés conformément : Au protocole Aux Procédures
CE QU IL FAUT SAVOIR PARTICIPATION À UN ESSAI CLINIQUE SUR UN MÉDICAMENT
 CE QU IL FAUT SAVOIR PARTICIPATION À UN ESSAI CLINIQUE SUR UN MÉDICAMENT Sommaire Comment se fait la recherche sur un nouveau médicament? (page 1) A quoi sert la recherche sur un nouveau médicament? (page
CE QU IL FAUT SAVOIR PARTICIPATION À UN ESSAI CLINIQUE SUR UN MÉDICAMENT Sommaire Comment se fait la recherche sur un nouveau médicament? (page 1) A quoi sert la recherche sur un nouveau médicament? (page
Responsabilité du promoteur et obligations des soustraitants. cliniques : conformité aux Bonnes Pratiques Cliniques et point de vue de l inspection
 Responsabilité du promoteur et obligations des soustraitants dans les essais cliniques : conformité aux Bonnes Pratiques Cliniques et point de vue de l inspection Anne RAISON Chef de l Unité Inspection
Responsabilité du promoteur et obligations des soustraitants dans les essais cliniques : conformité aux Bonnes Pratiques Cliniques et point de vue de l inspection Anne RAISON Chef de l Unité Inspection
A. Protocole de recherche (ainsi que l abrégé en langue française)
") Commission d'éthique cantonale (VD) de la recherche sur l'être humain Av. de Chailly, 23, 1012 Lausanne Courriel : [email protected] 5.12.2013/mz Recommandations pour la soumission d un dossier Tous
Commission d'éthique cantonale (VD) de la recherche sur l'être humain Av. de Chailly, 23, 1012 Lausanne Courriel : [email protected] 5.12.2013/mz Recommandations pour la soumission d un dossier Tous
PARTICIPATION À UN ESSAI CLINIQUE SUR UN MÉDICAMENT CE QU IL FAUT SAVOIR
 PARTICIPATION À UN ESSAI CLINIQUE SUR UN MÉDICAMENT CE QU IL FAUT SAVOIR SOMMAIRE COMMENT SE FAIT LA RECHERCHE SUR UN NOUVEAU MÉDICAMENT?...p. 3 À QUOI SERT LA RECHERCHE?...p. 4 QUELLES SONT LES GARANTIES?...p.
PARTICIPATION À UN ESSAI CLINIQUE SUR UN MÉDICAMENT CE QU IL FAUT SAVOIR SOMMAIRE COMMENT SE FAIT LA RECHERCHE SUR UN NOUVEAU MÉDICAMENT?...p. 3 À QUOI SERT LA RECHERCHE?...p. 4 QUELLES SONT LES GARANTIES?...p.
ASSOCIATION MEDICALE MONDIALE DECLARATION D HELSINKI Principes éthiques applicables à la recherche médicale impliquant des êtres humains
 ASSOCIATION MEDICALE MONDIALE DECLARATION D HELSINKI Principes éthiques applicables à la recherche médicale impliquant des êtres humains Adoptée par la 18e Assemblée générale de l AMM, Helsinki, Finlande,
ASSOCIATION MEDICALE MONDIALE DECLARATION D HELSINKI Principes éthiques applicables à la recherche médicale impliquant des êtres humains Adoptée par la 18e Assemblée générale de l AMM, Helsinki, Finlande,
Liège, le 29 juillet 2013. APPEL INTERNE et EXTERNE AUX CANDIDATURES N 2013-085
 Centre Hospitalier Universitaire de Liège Domaine Universitaire du Sart Tilman B35 4000 LIEGE 1 www.chuliege.be Département de Gestion des Ressources Humaines Service Recrutement Liège, le 29 juillet 2013
Centre Hospitalier Universitaire de Liège Domaine Universitaire du Sart Tilman B35 4000 LIEGE 1 www.chuliege.be Département de Gestion des Ressources Humaines Service Recrutement Liège, le 29 juillet 2013
Spécificité des essais cliniques dans le cadre de l enregistrement d un médicament. Florence BOUDEVIN 20 Novembre 2009
 Spécificité des essais cliniques dans le cadre de l enregistrement d un médicament Florence BOUDEVIN 20 Novembre 2009 Introduction Une étude clinique c est : Une molécule Un objectif principal Un cycle
Spécificité des essais cliniques dans le cadre de l enregistrement d un médicament Florence BOUDEVIN 20 Novembre 2009 Introduction Une étude clinique c est : Une molécule Un objectif principal Un cycle
Rôle de l ARCl. V Grimaud - UE recherche clinique - 18 mars 2011. Définitions
 Rôle de l ARCl 1 Définitions ARC : Assistant de Recherche Clinique TEC : Technicien d Étude Clinique Promoteur : Personne physique ou morale qui prend l initiative de la recherche Investigateur principal
Rôle de l ARCl 1 Définitions ARC : Assistant de Recherche Clinique TEC : Technicien d Étude Clinique Promoteur : Personne physique ou morale qui prend l initiative de la recherche Investigateur principal
Conduite des Essais Cliniques en Pharmacie Hospitalière Organisation du circuit du médicament
 Conduite des Essais Cliniques en Pharmacie Hospitalière Organisation du circuit du médicament Anne Daguenel-Nguyen Pharmacien Hôpital Saint-Antoine FARC 1 PREMIERE PARTIE: Quel est le rôle du pharmacien
Conduite des Essais Cliniques en Pharmacie Hospitalière Organisation du circuit du médicament Anne Daguenel-Nguyen Pharmacien Hôpital Saint-Antoine FARC 1 PREMIERE PARTIE: Quel est le rôle du pharmacien
La Gestion des Données Cliniques
 La Gestion des Données Cliniques Khaled Mostaguir, Ph.D, [email protected] Centre de Recherche Clinique HUG http://crc.hug-ge.ch/ La gestion des données clinique Le gestion des données au sein
La Gestion des Données Cliniques Khaled Mostaguir, Ph.D, [email protected] Centre de Recherche Clinique HUG http://crc.hug-ge.ch/ La gestion des données clinique Le gestion des données au sein
Item 169 : Évaluation thérapeutique et niveau de preuve
 Item 169 : Évaluation thérapeutique et niveau de preuve COFER, Collège Français des Enseignants en Rhumatologie Date de création du document 2010-2011 Table des matières ENC :...3 SPECIFIQUE :...3 I Différentes
Item 169 : Évaluation thérapeutique et niveau de preuve COFER, Collège Français des Enseignants en Rhumatologie Date de création du document 2010-2011 Table des matières ENC :...3 SPECIFIQUE :...3 I Différentes
Procédure normalisée de fonctionnement du RCBT Demande d informations additionnelles. 2.1.003 Version
 Numéro de PNF: Remplace: Objet: Procédure normalisée de fonctionnement du RCBT Demande d informations additionnelles 2.1.003 Version Demande d informations additionnelles Date d entrée en vigueur Catégorie
Numéro de PNF: Remplace: Objet: Procédure normalisée de fonctionnement du RCBT Demande d informations additionnelles 2.1.003 Version Demande d informations additionnelles Date d entrée en vigueur Catégorie
Déclarations européennes de la pharmacie hospitalière
 Déclarations européennes de la pharmacie hospitalière Les pages qui suivent constituent les Déclarations européennes de la pharmacie hospitalière. Elles représentent l expression consensuelle de ce que
Déclarations européennes de la pharmacie hospitalière Les pages qui suivent constituent les Déclarations européennes de la pharmacie hospitalière. Elles représentent l expression consensuelle de ce que
COMPRENDRE LA RECHERCHE CLINIQUE
 COMPRENDRE LA RECHERCHE CLINIQUE Unité de Recherche Clinique Avec le soutien de Roche 2 3 Sommaire Avant-propos 3 Accueil 4 Généralités 6 1. Qu est-ce qu une étude clinique? 2. Pourquoi participer à une
COMPRENDRE LA RECHERCHE CLINIQUE Unité de Recherche Clinique Avec le soutien de Roche 2 3 Sommaire Avant-propos 3 Accueil 4 Généralités 6 1. Qu est-ce qu une étude clinique? 2. Pourquoi participer à une
Création de procédures inter-services pour la gestion des essais de phase I à l Institut Gustave Roussy
 Création de procédures inter-services pour la gestion des essais de phase I à l Institut Gustave Roussy A.A. MOUSSA D. SCHWOB Institut de cancérologie Gustave-Roussy 94805 Villejuif cedex - FRANCE Plan
Création de procédures inter-services pour la gestion des essais de phase I à l Institut Gustave Roussy A.A. MOUSSA D. SCHWOB Institut de cancérologie Gustave-Roussy 94805 Villejuif cedex - FRANCE Plan
Formation CTOM «Printemps 2013»
 Formation CTOM «Printemps 2013» Formation intensive avec mise en œuvre opérationnelle 105 heures de formation en centre Formation professionnelle qualifiante au métier de Clinical Trials Operations Manager
Formation CTOM «Printemps 2013» Formation intensive avec mise en œuvre opérationnelle 105 heures de formation en centre Formation professionnelle qualifiante au métier de Clinical Trials Operations Manager
RISK-BASED APPROACH IN CLINICAL TRIALS objectives historical and regulatory context. Valérie Journot INSERM, F-CRIN WP4d, Bordeaux
 RISK-BASED APPROACH IN CLINICAL TRIALS objectives historical and regulatory context Valérie Journot INSERM, F-CRIN WP4d, Bordeaux Risk Management in clinical trials from trial monitoring to global risk
RISK-BASED APPROACH IN CLINICAL TRIALS objectives historical and regulatory context Valérie Journot INSERM, F-CRIN WP4d, Bordeaux Risk Management in clinical trials from trial monitoring to global risk
Pharmacovigilance des Essais cliniques
 Pharmacovigilance des Essais cliniques Edouard LECHAPTOIS DIU FIEC 22 janvier 2013 1 Introduction S o m m a i r e Eléments de réglementation Définitions Responsabilité des investigateurs / promoteurs Organisation
Pharmacovigilance des Essais cliniques Edouard LECHAPTOIS DIU FIEC 22 janvier 2013 1 Introduction S o m m a i r e Eléments de réglementation Définitions Responsabilité des investigateurs / promoteurs Organisation
Essais cliniques de phase 0 : état de la littérature 2006-2009
 17 èmes Journées des Statisticiens des Centres de Lutte contre le Cancer 4 ème Conférence Francophone d Epidémiologie Clinique Essais cliniques de phase 0 : état de la littérature 2006-2009 Q Picat, N
17 èmes Journées des Statisticiens des Centres de Lutte contre le Cancer 4 ème Conférence Francophone d Epidémiologie Clinique Essais cliniques de phase 0 : état de la littérature 2006-2009 Q Picat, N
ELABORATION DU PLAN DE MONITORING ADAPTE POUR UNE RECHERCHE BIOMEDICALE A PROMOTION INSTITUTIONNELLE
 Référence HCL : Titre de l étude : ELABORATION DU PLAN DE MONITORING ADAPTE POUR UNE RECHERCHE BIOMEDICALE A PROMOTION INSTITUTIONNELLE Investigateur Coordonnateur : Méthode. Définition du niveau de risque
Référence HCL : Titre de l étude : ELABORATION DU PLAN DE MONITORING ADAPTE POUR UNE RECHERCHE BIOMEDICALE A PROMOTION INSTITUTIONNELLE Investigateur Coordonnateur : Méthode. Définition du niveau de risque
secutrial Gestion des données d'études cliniques Les Colloques de l'uic Novembre 2012 Khaled Mostaguir, Ph.D, khaled.mostaguir@hcuge.
 secutrial Gestion des données d'études cliniques Les Colloques de l'uic Novembre 2012 Khaled Mostaguir, Ph.D, [email protected] Unité d Investigation Clinique Centre de Recherche Clinique HUG http://crc.hug-ge.ch/
secutrial Gestion des données d'études cliniques Les Colloques de l'uic Novembre 2012 Khaled Mostaguir, Ph.D, [email protected] Unité d Investigation Clinique Centre de Recherche Clinique HUG http://crc.hug-ge.ch/
QU EST-CE QU UNE INSPECTION? COMMENT S Y PREPARER?
 QU EST-CE QU UNE INSPECTION? COMMENT S Y PREPARER? DIU Formation des Attachés de Recherche Clinique - Techniciens d Essais Cliniques CHU Saint Antoine, 17 Décembre 2013 Dorian di-betta, R&D Opérations
QU EST-CE QU UNE INSPECTION? COMMENT S Y PREPARER? DIU Formation des Attachés de Recherche Clinique - Techniciens d Essais Cliniques CHU Saint Antoine, 17 Décembre 2013 Dorian di-betta, R&D Opérations
DEVELOPPEMENT DU MEDICAMENT 4 ème, 5 ème et 6 ème année de pharmacie
 DEVELOPPEMENT DU MEDICAMENT 4 ème, 5 ème et 6 ème année de pharmacie Nous proposons de nombreux stages dans différents domaines: Développement Cliniques, Affaires Réglementaires, Assurance Qualité, Gestion
DEVELOPPEMENT DU MEDICAMENT 4 ème, 5 ème et 6 ème année de pharmacie Nous proposons de nombreux stages dans différents domaines: Développement Cliniques, Affaires Réglementaires, Assurance Qualité, Gestion
Associé de recherche clinique
 Pharmabio Développement Associé de recherche clinique PROFIL DE COMPÉTENCES Mireille Lehoux, consultante Septembre 2009 ÉQUIPE DE PRODUCTION Coordination Francine Gendron Directrice générale Pharmabio
Pharmabio Développement Associé de recherche clinique PROFIL DE COMPÉTENCES Mireille Lehoux, consultante Septembre 2009 ÉQUIPE DE PRODUCTION Coordination Francine Gendron Directrice générale Pharmabio
Référentiel Officine
 Référentiel Officine Inscrire la formation dans la réalité et les besoins de la pharmacie d officine de demain - Ce référentiel décrit dans le cadre des missions et des activités du pharmacien d officine
Référentiel Officine Inscrire la formation dans la réalité et les besoins de la pharmacie d officine de demain - Ce référentiel décrit dans le cadre des missions et des activités du pharmacien d officine
OUTIL D'EVALUATION DU TEMPS ARC / CHEF DE PROJET PROMOTEUR REQUIS POUR UNE RECHERCHE BIOMEDICALE V 2.3 DE L OUTIL NOTICE D UTILISATION
 OUTIL D'EVALUATION DU TEMPS ARC / CHEF DE PROJET PROMOTEUR REQUIS POUR UNE RECHERCHE BIOMEDICALE V 2.3 DE L OUTIL NOTICE D UTILISATION i) Contexte :... - 2 - ii) But de l outil :... - 2 - iii) Fonctionnement
OUTIL D'EVALUATION DU TEMPS ARC / CHEF DE PROJET PROMOTEUR REQUIS POUR UNE RECHERCHE BIOMEDICALE V 2.3 DE L OUTIL NOTICE D UTILISATION i) Contexte :... - 2 - ii) But de l outil :... - 2 - iii) Fonctionnement
LA RECHERCHE CLINIQUE
 LA RECHERCHE CLINIQUE De la découverte scientifique à la mise à disposition d'un produit sur le marché (médicament, vaccin, outil diagnostic, dispositif médical...), il se passe plusieurs années, environ
LA RECHERCHE CLINIQUE De la découverte scientifique à la mise à disposition d'un produit sur le marché (médicament, vaccin, outil diagnostic, dispositif médical...), il se passe plusieurs années, environ
Dakar, Sénégal 5-9 Mars 2006. Dr Joël Keravec MSH/RPM Plus - Brésil et représentant le Globa
 éminaire pour les onsultants Francophones - estion des Approvisionments et des Stocks pour VIH, la TB et le Paludisme Assurance Qualité des Médicament s Dakar, Sénégal 5-9 Mars 2006 Dr Joël Keravec MSH/RPM
éminaire pour les onsultants Francophones - estion des Approvisionments et des Stocks pour VIH, la TB et le Paludisme Assurance Qualité des Médicament s Dakar, Sénégal 5-9 Mars 2006 Dr Joël Keravec MSH/RPM
ANNEXES AU REGLEMENT N 06/2010/CM/UEMOA
 UNION ECONOMIQUE ET MONETAIRE OUEST AFRICAINE ------------------------- La Commission ANNEXES AU REGLEMENT N 06/2010/CM/UEMOA LES ANNEXES AU REGLEMENT RELATIF AUX PROCEDURES D HOMOLOGATION DES PRODUITS
UNION ECONOMIQUE ET MONETAIRE OUEST AFRICAINE ------------------------- La Commission ANNEXES AU REGLEMENT N 06/2010/CM/UEMOA LES ANNEXES AU REGLEMENT RELATIF AUX PROCEDURES D HOMOLOGATION DES PRODUITS
M.Benmimoun MD,MBA Medical Operations Director
 M.Benmimoun MD,MBA Medical Operations Director Vos Responsabilités en Matière de Pharmacovigilance Notification Spontanée d'événements Indésirables Formation 2 Notification Spontanée d Événement Indésirable
M.Benmimoun MD,MBA Medical Operations Director Vos Responsabilités en Matière de Pharmacovigilance Notification Spontanée d'événements Indésirables Formation 2 Notification Spontanée d Événement Indésirable
Un métier en évolution pour répondre aux nouvelles. Face à ces évolutions, un nouveau métier
 Les métiers de la pharmacovigilance Des métiers en évolution dans une industrie de haute technologie et d'innovation Une mission d'évaluation et de gestion des risques liés à l'utilisation des médicaments
Les métiers de la pharmacovigilance Des métiers en évolution dans une industrie de haute technologie et d'innovation Une mission d'évaluation et de gestion des risques liés à l'utilisation des médicaments
Partie 1. Principes. Karmela Krleža-Jerić, An-Wen Chan, Kay Dickersin, Ida Sim, Jeremy Grimshaw, Christian Gluud, for the Ottawa GroupT 1
 Déclaration d Ottawa sur l enregistrement des essais d interventions de santé: Proposition pour l enregistrement international d informations relatives au protocole et de résultats des essais réalisés
Déclaration d Ottawa sur l enregistrement des essais d interventions de santé: Proposition pour l enregistrement international d informations relatives au protocole et de résultats des essais réalisés
Encadrement réglementaire
 Méthodologie de la Recherche Clinique [email protected] Encadrement réglementaire Rappel des méthodologies d'études Parcours chercheur en réponse aux appels d'offres Recherche documentaire
Méthodologie de la Recherche Clinique [email protected] Encadrement réglementaire Rappel des méthodologies d'études Parcours chercheur en réponse aux appels d'offres Recherche documentaire
1 OBJET DE LA PROCÉDURE
 1 OBJET DE LA PROCÉDURE La procédure décrit comment soumettre une expérimentation médicale dont l investigateur est cadre médical permanent des CUSL au Comité d Ethique Hospitalo-Facultaire pour avis.
1 OBJET DE LA PROCÉDURE La procédure décrit comment soumettre une expérimentation médicale dont l investigateur est cadre médical permanent des CUSL au Comité d Ethique Hospitalo-Facultaire pour avis.
La politique pharmaceutique à l hôpital (PPH) : élémentaire pour la dispense globale de soins
 : élémentaire pour la dispense globale de soins") La politique pharmaceutique à l hôpital (PPH) : élémentaire pour la dispense globale de soins Eléments pour une note de politique Introduction Les médicaments prennent une place de plus en plus importante
La politique pharmaceutique à l hôpital (PPH) : élémentaire pour la dispense globale de soins Eléments pour une note de politique Introduction Les médicaments prennent une place de plus en plus importante
Répondre à un appel à projet - Vie d un projet
 Répondre à un appel à projet - Vie d un projet Carine Coffre 1, Nathalie Juteau 1, Ken Haguenoer 2,3 16 décembre 2011 1. Centre d Investigation Clinique - Inserm 202 CHRU de Tours 2. Laboratoire de Santé
Répondre à un appel à projet - Vie d un projet Carine Coffre 1, Nathalie Juteau 1, Ken Haguenoer 2,3 16 décembre 2011 1. Centre d Investigation Clinique - Inserm 202 CHRU de Tours 2. Laboratoire de Santé
Article 1 er. Code de la santé publique Texte précédent. Nouveau texte. 28/11/2012 Evelyne Pierron Consultants. Article R5121-25
 Tableau comparatif Décret n 2012-1244 du 8 novembre 2012 relatif au renforcement des dispositions en matière de sécurité des médicaments à usage humain soumis à autorisation de mise sur le marché et à
Tableau comparatif Décret n 2012-1244 du 8 novembre 2012 relatif au renforcement des dispositions en matière de sécurité des médicaments à usage humain soumis à autorisation de mise sur le marché et à
parer à une inspection
 Comment se préparer parer à une inspection DIU FARC Master BIP Licence professionnelle des métiers de la recherche clinique Hôpital Saint Antoine - 15 Décembre 2010 Philippe Enfrin Opérations Scientifiques
Comment se préparer parer à une inspection DIU FARC Master BIP Licence professionnelle des métiers de la recherche clinique Hôpital Saint Antoine - 15 Décembre 2010 Philippe Enfrin Opérations Scientifiques
L application doit être validée et l infrastructure informatique doit être qualifiée.
 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 Annexe 11: Systèmes informatisés
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 Annexe 11: Systèmes informatisés
«Adaptation de la mise en œuvre des bonnes pratiques cliniques en fonction des caractéristiques de certaines recherches»
 Synthèse de la table ronde 2- Giens XXI -octobre 2005 «Adaptation de la mise en œuvre des bonnes pratiques cliniques en fonction des caractéristiques de certaines recherches» Pierre-Henri.Bertoye, Soizic.Courcier-Duplantier,
Synthèse de la table ronde 2- Giens XXI -octobre 2005 «Adaptation de la mise en œuvre des bonnes pratiques cliniques en fonction des caractéristiques de certaines recherches» Pierre-Henri.Bertoye, Soizic.Courcier-Duplantier,
QUESTION D ETHIQUE APPROCHE ENVIRONNEMENT- SANTE. 10ème cours d été 2006 Bamako SIFEE Pr HOUENOU AGBO Yveline UFR Sciences Médicales d Abidjan
 QUESTION D ETHIQUE APPROCHE ENVIRONNEMENT- SANTE 10ème cours d été 2006 Bamako SIFEE Pr HOUENOU AGBO Yveline UFR Sciences Médicales d Abidjan INTRODUCTION Environnement et Santé Évaluation environnementale
QUESTION D ETHIQUE APPROCHE ENVIRONNEMENT- SANTE 10ème cours d été 2006 Bamako SIFEE Pr HOUENOU AGBO Yveline UFR Sciences Médicales d Abidjan INTRODUCTION Environnement et Santé Évaluation environnementale
Foire Aux Questions. Sur les recherches biomédicales
 Foire Aux Questions Sur les recherches biomédicales Version 5.0 (Octobre 2011) Historique du document Date de publication 19 octobre 2011 07 mai 2012 (suite à la création de l ANSM en remplacement de l
Foire Aux Questions Sur les recherches biomédicales Version 5.0 (Octobre 2011) Historique du document Date de publication 19 octobre 2011 07 mai 2012 (suite à la création de l ANSM en remplacement de l
Pour les utilisateurs Investigateurs et patients. Philadelphia, PA / Gif sur Yvette, France Mai 2010
 Collecter les données patients avec une solution de capture de données électroniques basée sur du papier: la meilleure solution pour garantir la plus haute qualité des données et préserver une donnée source
Collecter les données patients avec une solution de capture de données électroniques basée sur du papier: la meilleure solution pour garantir la plus haute qualité des données et préserver une donnée source
LIGNES DIRECTRICES CLINIQUES TOUT AU LONG DU CONTINUUM DE SOINS : Objectif de ce chapitre. 6.1 Introduction 86
 LIGNES DIRECTRICES CLINIQUES TOUT AU LONG DU CONTINUUM DE SOINS : ÉTABLISSEMENT DE LIENS ENTRE LES PERSONNES CHEZ QUI UN DIAGNOSTIC D INFECTION À VIH A ÉTÉ POSÉ ET LES SERVICES DE SOINS ET DE TRAITEMENT
LIGNES DIRECTRICES CLINIQUES TOUT AU LONG DU CONTINUUM DE SOINS : ÉTABLISSEMENT DE LIENS ENTRE LES PERSONNES CHEZ QUI UN DIAGNOSTIC D INFECTION À VIH A ÉTÉ POSÉ ET LES SERVICES DE SOINS ET DE TRAITEMENT
«Quelle information aux patients en recherche biomédicale? Quels enseignements en retirer pour la pratique quotidienne?»
 «Quelle information aux patients en recherche biomédicale? Quels enseignements en retirer pour la pratique quotidienne?» Dr Adeline Paris Unité de Pharmacologie Clinique Centre d Investigation Clinique
«Quelle information aux patients en recherche biomédicale? Quels enseignements en retirer pour la pratique quotidienne?» Dr Adeline Paris Unité de Pharmacologie Clinique Centre d Investigation Clinique
Gestion des bases de données
 Gestion des bases de données DU Chef de Projet en Recherche Clinique 23/11/2012 Fabrice GOURMELON URC/CIC Necker - Cochin 2 A. Qu est-ce qu une donnée? B. Qu est-ce qu une base de données? C. Définition
Gestion des bases de données DU Chef de Projet en Recherche Clinique 23/11/2012 Fabrice GOURMELON URC/CIC Necker - Cochin 2 A. Qu est-ce qu une donnée? B. Qu est-ce qu une base de données? C. Définition
Guide de rédaction d un protocole de recherche clinique à. l intention des chercheurs évoluant en recherche fondamentale
 V E R S I O N A V R I L 2 0 1 2 C E N T R E D E R E C H E R C H E C L I N I Q U E É T I E N N E - L E B E L D U C H U S Guide de rédaction d un protocole de recherche clinique à l intention des chercheurs
V E R S I O N A V R I L 2 0 1 2 C E N T R E D E R E C H E R C H E C L I N I Q U E É T I E N N E - L E B E L D U C H U S Guide de rédaction d un protocole de recherche clinique à l intention des chercheurs
Montréal, 24 mars 2015. David Levine Président et chef de la direction DL Strategic Consulting. DL Consulting Strategies in Healthcare
 Montréal, 24 mars 2015 David Levine Président et chef de la direction DL Strategic Consulting 1 RSSPQ, 2013 2 MÉDECINE INDIVIDUALISÉE Médecine personnalisée Médecine de précision Biomarqueurs Génomique
Montréal, 24 mars 2015 David Levine Président et chef de la direction DL Strategic Consulting 1 RSSPQ, 2013 2 MÉDECINE INDIVIDUALISÉE Médecine personnalisée Médecine de précision Biomarqueurs Génomique
Les systèmes CDMS. et les logiciels EDC
 Les systèmes CDMS et les logiciels EDC Khaled Mostaguir, Ph.D, [email protected] Centre de Recherche Clinique HUG http://crc.hug-ge.ch/ Les systèmes CDMS et les logiciels EDC Les systèmes CDMS
Les systèmes CDMS et les logiciels EDC Khaled Mostaguir, Ph.D, [email protected] Centre de Recherche Clinique HUG http://crc.hug-ge.ch/ Les systèmes CDMS et les logiciels EDC Les systèmes CDMS
GUIDE POUR LA MISE SUR LE MARCHÉ DE DISPOSITIFS MÉDICAUX SUR MESURE APPLIQUE AU SECTEUR DENTAIRE
 Actualisation Mai 2012 Direction de l'evaluation des Dispositifs Médicaux Département Surveillance du Marché GUIDE POUR LA MISE SUR LE MARCHÉ DE DISPOSITIFS MÉDICAUX SUR MESURE APPLIQUE AU SECTEUR DENTAIRE
Actualisation Mai 2012 Direction de l'evaluation des Dispositifs Médicaux Département Surveillance du Marché GUIDE POUR LA MISE SUR LE MARCHÉ DE DISPOSITIFS MÉDICAUX SUR MESURE APPLIQUE AU SECTEUR DENTAIRE
Master UP 6. Mention Santé Publique et Management de la Santé. Spécialité Pharmacologie Clinique. Construire une carrière dans l industrie
 Master UP 6 Mention Santé Publique et Management de la Santé Spécialité Pharmacologie Clinique Construire une carrière dans l industrie pharmaceutique Alain Leclerc, CTPartners 3 mars 2009 Your Executive
Master UP 6 Mention Santé Publique et Management de la Santé Spécialité Pharmacologie Clinique Construire une carrière dans l industrie pharmaceutique Alain Leclerc, CTPartners 3 mars 2009 Your Executive
admission directe du patient en UNV ou en USINV
 Société française de neurologie RÉFÉRENTIEL D AUTO-ÉVALUATION DES PRATIQUES EN NEUROLOGIE Prise en charge hospitalière initiale des personnes ayant fait un accident vasculaire cérébral (AVC) : admission
Société française de neurologie RÉFÉRENTIEL D AUTO-ÉVALUATION DES PRATIQUES EN NEUROLOGIE Prise en charge hospitalière initiale des personnes ayant fait un accident vasculaire cérébral (AVC) : admission
ACTUALITES THERAPEUTIQUES. Dr Sophie PITTION (CHU Nancy) Metz, le 2 Juin 2012
 Metz, le 2 Juin 2012") ACTUALITES THERAPEUTIQUES Dr Sophie PITTION (CHU Nancy) Metz, le 2 Juin 2012 Traitement de fond Objectifs: Réduire le nombre de poussées Arrêter ou freiner la progression du handicap Les traitements disponibles
ACTUALITES THERAPEUTIQUES Dr Sophie PITTION (CHU Nancy) Metz, le 2 Juin 2012 Traitement de fond Objectifs: Réduire le nombre de poussées Arrêter ou freiner la progression du handicap Les traitements disponibles
SECTION II RELATIVE AU PRÉLEVEUR
 SECTION II RELATIVE AU PRÉLEVEUR II-0 INDEX SECTION II Pages Section relative au préleveur Heures d ouvertures des laboratoires pour clients externes Requête régionale II-2 II-2 II-3 Informations requises
SECTION II RELATIVE AU PRÉLEVEUR II-0 INDEX SECTION II Pages Section relative au préleveur Heures d ouvertures des laboratoires pour clients externes Requête régionale II-2 II-2 II-3 Informations requises
Guide à l intention des patients sur les thérapies à base de cellules souches
 Guide à l intention des patients sur les thérapies à base de cellules souches Appendice I des Lignes directrices pour l application en clinique des cellules souches Traduction fournie par le Réseau de
Guide à l intention des patients sur les thérapies à base de cellules souches Appendice I des Lignes directrices pour l application en clinique des cellules souches Traduction fournie par le Réseau de
admission aux urgences
 Société française de neurologie RÉFÉRENTIEL D AUTO-ÉVALUATION DES PRATIQUES EN NEUROLOGIE Prise en charge hospitalière initiale des personnes ayant fait un accident vasculaire cérébral (AVC) : admission
Société française de neurologie RÉFÉRENTIEL D AUTO-ÉVALUATION DES PRATIQUES EN NEUROLOGIE Prise en charge hospitalière initiale des personnes ayant fait un accident vasculaire cérébral (AVC) : admission
Médicaments en vente libre : considérations pour la pratique de la physiothérapie
 Médicaments en vente libre : considérations pour la pratique de la physiothérapie Adapté d un article approuvé de l Alliance canadienne des organismes de réglementation de la physiothérapie (2012) Le CPTNB
Médicaments en vente libre : considérations pour la pratique de la physiothérapie Adapté d un article approuvé de l Alliance canadienne des organismes de réglementation de la physiothérapie (2012) Le CPTNB
Guide pratique sur l'encadrement de la recherche biomédicale. La protection des droits de la personne
 Guide pratique sur l'encadrement de la recherche biomédicale Dispositions législatives relatives au chapitre : La protection des droits de la personne Code de la santé publique Dispositions introduites
Guide pratique sur l'encadrement de la recherche biomédicale Dispositions législatives relatives au chapitre : La protection des droits de la personne Code de la santé publique Dispositions introduites
LE CHEMINEMENT COMPLEXE D UN VACCIN
 LE CHEMINEMENT COMPLEXE D UN VACCIN Chaîne de production, exigences réglementaires et accès aux vaccins International Federation of Pharmaceutical Manufacturers & Associations LE CHEMINEMENT COMPLEXE D
LE CHEMINEMENT COMPLEXE D UN VACCIN Chaîne de production, exigences réglementaires et accès aux vaccins International Federation of Pharmaceutical Manufacturers & Associations LE CHEMINEMENT COMPLEXE D
RECOMMANDATIONS DU COLLEGE A PROPOS DU PHARMACIEN ADJOINT MAITRE DE STAGE ADJOINT
 RECOMMANDATIONS DU COLLEGE A PROPOS DU PHARMACIEN ADJOINT MAITRE DE STAGE ADJOINT L agrément de maître de stage repose à la fois sur des critères liés à l officine d une part et à son titulaire d autre
RECOMMANDATIONS DU COLLEGE A PROPOS DU PHARMACIEN ADJOINT MAITRE DE STAGE ADJOINT L agrément de maître de stage repose à la fois sur des critères liés à l officine d une part et à son titulaire d autre
Ministère des Affaires Étrangères et de la Coopération Internationale. Programme d appui au secteur de la santé- 8 ÈME FED
 République du Mali Ministère des Affaires Étrangères et de la Coopération Internationale Programme d appui au secteur de la santé- 8 ÈME FED Mars 2006 Elaboration d un guide concernant les grossistes sur
République du Mali Ministère des Affaires Étrangères et de la Coopération Internationale Programme d appui au secteur de la santé- 8 ÈME FED Mars 2006 Elaboration d un guide concernant les grossistes sur
Activité des programmes de médicaments
 Chapitre 4 Section 4.05 Ministère de la Santé et des Soins de longue durée Activité des programmes de médicaments Suivi des vérifications de l optimisation des ressources, section 3.05 du Rapport annuel
Chapitre 4 Section 4.05 Ministère de la Santé et des Soins de longue durée Activité des programmes de médicaments Suivi des vérifications de l optimisation des ressources, section 3.05 du Rapport annuel
Table des matières chronologique volume 1 médicaments
 volume 1 médicaments vii Lois - versions consolidées Loi du 15 mai 2007 relative à la répression de la contrefaçon et de la piraterie de droits de propriété intellectuelle........... 3 Chapitre I. Disposition
volume 1 médicaments vii Lois - versions consolidées Loi du 15 mai 2007 relative à la répression de la contrefaçon et de la piraterie de droits de propriété intellectuelle........... 3 Chapitre I. Disposition
Florence DEFRESNE, Dominique VAN OPHEM
 Procédure de déclaration des faits de non conformité, déviation, violation et problèmes inattendus lors d'une étude clinique N :AAHRPP-SOP-040 / REV Date d application : 24/04/2014 002 Rédigé par : Relu
Procédure de déclaration des faits de non conformité, déviation, violation et problèmes inattendus lors d'une étude clinique N :AAHRPP-SOP-040 / REV Date d application : 24/04/2014 002 Rédigé par : Relu
3 Guide pour développer un plan national de gestion des déchets de soins médicaux
 3 Guide pour développer un plan national de gestion des déchets de soins médicaux (111) Cette section présente une liste d actions recommandées qui devraient être mises en place par le gouvernement central
3 Guide pour développer un plan national de gestion des déchets de soins médicaux (111) Cette section présente une liste d actions recommandées qui devraient être mises en place par le gouvernement central
La recherche clinique de demain ne se fera pas sans les paramédicaux
 La recherche clinique de demain ne se fera pas sans les paramédicaux Marc Beaumont, kinésithérapeute - 5ème journée inter régionale GIRCI - Tours, 3 juin 2015 Qu est ce que la recherche clinique? «une
La recherche clinique de demain ne se fera pas sans les paramédicaux Marc Beaumont, kinésithérapeute - 5ème journée inter régionale GIRCI - Tours, 3 juin 2015 Qu est ce que la recherche clinique? «une
Risques et dispositifs médicaux. «Responsabilités encourues» Isabelle Lucas-Baloup. 12, 13 et 14 octobre 2010
 Risques et dispositifs médicaux «Responsabilités encourues» 1 Le circuit du dispositif médical Responsabilité D.M. approche systématique approche du produit implique analyse des missions et responsabilités
Risques et dispositifs médicaux «Responsabilités encourues» 1 Le circuit du dispositif médical Responsabilité D.M. approche systématique approche du produit implique analyse des missions et responsabilités
Information sur les programmes d autorisation préalable, de pharmacie désignée et de gestion des dossiers médicaux. Autorisation préalable
 Information sur les programmes d autorisation préalable, de pharmacie désignée et de gestion des dossiers médicaux La présente feuille de renseignements vise à fournir de l information sur le processus
Information sur les programmes d autorisation préalable, de pharmacie désignée et de gestion des dossiers médicaux La présente feuille de renseignements vise à fournir de l information sur le processus
EXIGENCES MINIMALES RELATIVES À LA PROTECTION DES RENSEIGNEMENTS PERSONNELS LORS DE SONDAGES RÉALISÉS PAR UN ORGANISME PUBLIC OU SON MANDATAIRE
 EXIGENCES MINIMALES RELATIVES À LA PROTECTION DES RENSEIGNEMENTS PERSONNELS LORS DE SONDAGES RÉALISÉS PAR UN ORGANISME PUBLIC OU SON MANDATAIRE JUIN 1999 Exigences minimales relatives à la protection des
EXIGENCES MINIMALES RELATIVES À LA PROTECTION DES RENSEIGNEMENTS PERSONNELS LORS DE SONDAGES RÉALISÉS PAR UN ORGANISME PUBLIC OU SON MANDATAIRE JUIN 1999 Exigences minimales relatives à la protection des
«INNOVATION PEDAGOGIQUE PAR LA MISE EN PLACE D UNE UNITE DE PHARMACIE EXPERIMENTALE POUR AMELIORER LA QUALITE DE LA FORMATION PROFESSIONNALISANTE»
 REPUBLIQUE TUNISIENNE MINISTERE DE L'ENSEIGNEMENT SUPERIEUR DE LA RECHERCHE SCIENTIFIQUE Université de Monastir Faculté de pharmacie de Monastir «INNOVATION PEDAGOGIQUE PAR LA MISE EN PLACE D UNE UNITE
REPUBLIQUE TUNISIENNE MINISTERE DE L'ENSEIGNEMENT SUPERIEUR DE LA RECHERCHE SCIENTIFIQUE Université de Monastir Faculté de pharmacie de Monastir «INNOVATION PEDAGOGIQUE PAR LA MISE EN PLACE D UNE UNITE
NOTE DE SYNTHESE RELATIVE AUX COMMENTAIRES SUR LE PROJET DE DECRET N 2-14-841 RELATIF A L AUTORISATION DE MISE SUR LE MARCHE DES MEDICAMENTS A USAGE
 NOTE DE SYNTHESE RELATIVE AUX COMMENTAIRES SUR LE PROJET DE DECRET N 2-14-841 RELATIF A L AUTORISATION DE MISE SUR LE MARCHE DES MEDICAMENTS A USAGE HUMAIN AVEC LEURS REPONSES CORRESPONDANTES 1 Mme S.
NOTE DE SYNTHESE RELATIVE AUX COMMENTAIRES SUR LE PROJET DE DECRET N 2-14-841 RELATIF A L AUTORISATION DE MISE SUR LE MARCHE DES MEDICAMENTS A USAGE HUMAIN AVEC LEURS REPONSES CORRESPONDANTES 1 Mme S.
Conclusions du Conseil sur l'innovation dans l'intérêt des patients
 Conseil de l'union Européenne PRESSE FR CONCLUSIONS DU CONSEIL Bruxelles, le 1 décembre 2014 Conclusions du Conseil sur l'innovation dans l'intérêt des patients Session du Conseil Emploi, politique sociale,
Conseil de l'union Européenne PRESSE FR CONCLUSIONS DU CONSEIL Bruxelles, le 1 décembre 2014 Conclusions du Conseil sur l'innovation dans l'intérêt des patients Session du Conseil Emploi, politique sociale,
A009 Maîtrise des enregistrements
 28.11.2014 Version 11 Page 1 de 8 A009 Maîtrise des enregistrements Modifications : pages 2, 5 South Lane Tower I 1, avenue du Swing L-4367 Belvau Tél.: (+352) 2477 4360 Fa: (+352) 2479 4360 [email protected]
28.11.2014 Version 11 Page 1 de 8 A009 Maîtrise des enregistrements Modifications : pages 2, 5 South Lane Tower I 1, avenue du Swing L-4367 Belvau Tél.: (+352) 2477 4360 Fa: (+352) 2479 4360 [email protected]
Appel à Manifestation d'intérêt
 AfricaInteract : Renforcement des liens entre la recherche et les décideurs politiques pour l'adaptation au changement climatique en Afrique Appel à Manifestation d'intérêt Recrutement d'un expert pour
AfricaInteract : Renforcement des liens entre la recherche et les décideurs politiques pour l'adaptation au changement climatique en Afrique Appel à Manifestation d'intérêt Recrutement d'un expert pour
étude de fonctions rémunérations Industrie du médicament
 2011 2012 étude de fonctions rémunérations Industrie du médicament & sommaire 3 Editorial Depuis plus d une décennie que Michael Page France intervient sur le marché pharmaceutique, il était grand temps
2011 2012 étude de fonctions rémunérations Industrie du médicament & sommaire 3 Editorial Depuis plus d une décennie que Michael Page France intervient sur le marché pharmaceutique, il était grand temps
Charte pour la communication sur internet des entreprises pharmaceutiques
 DIRECTION DE L EVALUATION DE LA PUBLICITE, DES PRODUITS COSMETIQUES ET DES BIOCIDES Charte pour la communication sur internet des entreprises pharmaceutiques Préambule Mise à jour 2010 Au plan mondial,
DIRECTION DE L EVALUATION DE LA PUBLICITE, DES PRODUITS COSMETIQUES ET DES BIOCIDES Charte pour la communication sur internet des entreprises pharmaceutiques Préambule Mise à jour 2010 Au plan mondial,
COMMENT MAITRISER LA GESTION DES APPROVISIONNEMENTS ET DES STOCKS DE MEDICAMENTS
 1 sur 9 COMMENT MAITRISER LA GESTION DES APPROVISIONNEMENTS ET DES STOCKS DE MEDICAMENTS (L'article intégral est paru dans Gestions Hospitalières n 357 de juin-juillet 1996) Pour plus d'informations concernant
1 sur 9 COMMENT MAITRISER LA GESTION DES APPROVISIONNEMENTS ET DES STOCKS DE MEDICAMENTS (L'article intégral est paru dans Gestions Hospitalières n 357 de juin-juillet 1996) Pour plus d'informations concernant
Diplôme d Etat d infirmier Référentiel de compétences
 Annexe II Diplôme d Etat d infirmier Référentiel de compétences Les référentiels d activités et de compétences du métier d infirmier diplômé d Etat ne se substituent pas au cadre réglementaire. En effet,
Annexe II Diplôme d Etat d infirmier Référentiel de compétences Les référentiels d activités et de compétences du métier d infirmier diplômé d Etat ne se substituent pas au cadre réglementaire. En effet,
Essais cliniques de médicaments : ce qui va changer. Dr Philippe VELLA Chef de l Unité Essais Cliniques
 Essais cliniques de médicaments : ce qui va changer Dr Philippe VELLA Chef de l Unité Essais Cliniques Besançon 25 novembre 2004 Essais cliniques de médicaments : ce qui va changer Début de l essai Procédure
Essais cliniques de médicaments : ce qui va changer Dr Philippe VELLA Chef de l Unité Essais Cliniques Besançon 25 novembre 2004 Essais cliniques de médicaments : ce qui va changer Début de l essai Procédure
EVALUATION DES TECHNOLOGIES DE SANTÉ ANALYSE MÉDICO-ÉCONOMIQUE. Efficacité et efficience des hypolipémiants Une analyse centrée sur les statines
 EVALUATION DES TECHNOLOGIES DE SANTÉ ANALYSE MÉDICO-ÉCONOMIQUE Efficacité et efficience des hypolipémiants Une analyse centrée sur les statines Juillet 2010 Mise à jour Septembre 2010 1 Le rapport complet
EVALUATION DES TECHNOLOGIES DE SANTÉ ANALYSE MÉDICO-ÉCONOMIQUE Efficacité et efficience des hypolipémiants Une analyse centrée sur les statines Juillet 2010 Mise à jour Septembre 2010 1 Le rapport complet
Evolution de la législation sur la recherche
 Evolution de la législation sur la recherche Pr Francois Lemaire SRLF 07-10-11 DRCD de l AP-HP, Saint-Louis La PPL Jardé Loi HPST : novembre 07 - septembre 08 (rapporteur pressenti: O Jardé) PPl Jardé
Evolution de la législation sur la recherche Pr Francois Lemaire SRLF 07-10-11 DRCD de l AP-HP, Saint-Louis La PPL Jardé Loi HPST : novembre 07 - septembre 08 (rapporteur pressenti: O Jardé) PPl Jardé
Révision des descriptions génériques Comment monter un dossier?
 DISPOSITIFS MEDICAUX Révision des descriptions génériques Comment monter un dossier? Guide pour le dossier déposé par les fabricants/distributeurs Adopté en séance de la CEPP* le 13 juillet 2005 *CEPP
DISPOSITIFS MEDICAUX Révision des descriptions génériques Comment monter un dossier? Guide pour le dossier déposé par les fabricants/distributeurs Adopté en séance de la CEPP* le 13 juillet 2005 *CEPP
Conseil et Etudes de Marché du pareil au même? Audrey Chemin [email protected] European Market Analytics Manager Oncology
 Conseil et Etudes de Marché du pareil au même? Audrey Chemin [email protected] European Market Analytics Manager Oncology Mon Curriculum Sept 09 - auj Sept 08 Aug 09 Sept 06 Aug 08 Sept 06 Feb 06
Conseil et Etudes de Marché du pareil au même? Audrey Chemin [email protected] European Market Analytics Manager Oncology Mon Curriculum Sept 09 - auj Sept 08 Aug 09 Sept 06 Aug 08 Sept 06 Feb 06
PROCEDURE DE CERTIFICATION IIW MCS SELON EN ISO 3834
 1 PROCEDURE DE CERTIFICATION IIW MCS SELON EN 2 SYNTHESE DES REVISIONS Révision 0 : Révision 1 : édition originale de la procédure modification du paragraphe 3.3.1 c) : critères pour évaluation des coordonnateurs
1 PROCEDURE DE CERTIFICATION IIW MCS SELON EN 2 SYNTHESE DES REVISIONS Révision 0 : Révision 1 : édition originale de la procédure modification du paragraphe 3.3.1 c) : critères pour évaluation des coordonnateurs
Gestion éthique des banques de recherche
 Gestion éthique des banques de recherche Jean-Marie Therrien, Ph.D. Président du Comité d éthique de la recherche, CHU Sainte-Justine 30 octobre 2012 Plan Justification éthique et scientifique Principes
Gestion éthique des banques de recherche Jean-Marie Therrien, Ph.D. Président du Comité d éthique de la recherche, CHU Sainte-Justine 30 octobre 2012 Plan Justification éthique et scientifique Principes
PARTENAIRE COMMERCIAL DE MSD CODE DE CONDUITE
 PARTENAIRE COMMERCIAL DE MSD CODE DE CONDUITE «Nos valeurs et nos règles» pour nos partenaires commerciaux Code de conduite pour le partenaire commercial de MSD [Édition 1] MSD est engagée dans une démarche
PARTENAIRE COMMERCIAL DE MSD CODE DE CONDUITE «Nos valeurs et nos règles» pour nos partenaires commerciaux Code de conduite pour le partenaire commercial de MSD [Édition 1] MSD est engagée dans une démarche
3 e symposium international
 3 e symposium international de l Association internationale des Acheteurs et Approvisionneurs publics et privés de la Santé Bruxelles, les 18 et 19 octobre 2012 Les Achats: Du support à la coordination
3 e symposium international de l Association internationale des Acheteurs et Approvisionneurs publics et privés de la Santé Bruxelles, les 18 et 19 octobre 2012 Les Achats: Du support à la coordination
LES BASES JURIDIQUES DE LA RESPONSABILITE & DE L ASSURANCE EN MATIERE DE RECHERCHE BIOMEDICALE DIU-FARC-TEC 04/11/2009 1
 LES BASES JURIDIQUES DE LA RESPONSABILITE & DE L ASSURANCE EN MATIERE DE RECHERCHE BIOMEDICALE DIU-FARC-TEC 04/11/2009 1 1ère Partie : La recherche biomédicale en France Historique Responsabilité et Assurance
LES BASES JURIDIQUES DE LA RESPONSABILITE & DE L ASSURANCE EN MATIERE DE RECHERCHE BIOMEDICALE DIU-FARC-TEC 04/11/2009 1 1ère Partie : La recherche biomédicale en France Historique Responsabilité et Assurance
intégration tri ogique Démarches Processus au service des enjeux de la Santé
 Démarches Processus au service des enjeux de la Santé Sommaire 1. Présentation d IDS Scheer et de ses Partenaires «Santé» 2. Modélisation par les métiers des processus et données clés dans le secteur «Santé»
Démarches Processus au service des enjeux de la Santé Sommaire 1. Présentation d IDS Scheer et de ses Partenaires «Santé» 2. Modélisation par les métiers des processus et données clés dans le secteur «Santé»